










Serviços Personalizados
Journal

Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares em
SciELO
Similares em
SciELO Compartilhar

 Permalink
PermalinkRevista de Ciências Agrárias
versão impressa ISSN 0871-018X
Rev. de Ciências Agrárias v.33 n.2 Lisboa dez. 2010
Phylogenetic diversity of methanogenic archae in diets with different hay proportions
Marta de Campos Neves1, Luciano Takeshi Kishi2, Eliane Cristina da Cunha Alves1, Jane Maria Bertocco Ezequiel3, Manoel Victor Franco Lemos1
1Universidade Estadual Paulista, Faculdade de Ciências Agrárias e Veterinárias, Departamento de Biologia Aplicada à Agropecuária, Via de Acesso Prof. Paulo Donato Castellane, s/no, CEP 14884-900, Jaboticabal, SP, Brasil. E-mail: martacamposneves@yahoo.com.br
2Universidade Estadual Paulista, Faculdade de Ciências Agrárias e Veterinárias, Departamento de Tecnologia.
3Universidade Estadual Paulista, Faculdade de Ciências Agrárias e Veterinárias, Departamento de Zootecnia.
ABSTRACT
The objective of this research was to detect the presence of archaea in the bovine rumen using genetic sequences from the conserved region of the 16S rDNA. Samples were collected from bovines that were fed two different experimental dietetic ratios of roughage to concentrate. The 16S rDNA region was amplified using PCR and analyzed using the Phred/Phrap/Consed programs. For the treatment with 70% hay diet 96 sequences related to the Methanobacteriaceae family, 47 sequences from non-cultured archaea, and 60 sequences from unknown archaea were identified by the BLAST analysis. For the treatment with 30% hay diet the BLAST analysis identified 125 sequences belonging to the Methanobacteriaceae fa-mily 42 sequences from non-cultured archaea, and 32 sequences from unknown archaea. The analysis of the 16S rDNA sequences of archaea collected from the bovine rumen, allows more sequences matching the unknown archaea were identified in the treatment with 70% hay.
Key-words: Bovine rumen, methanogens, molecular analysis, PCR.
Diversidade filogenética de archaea metanogênica em dietas com diferentes proporções de feno
RESUMO
O objetivo deste trabalho foi detectar a presença de arqueias no rúmen bovino por meio das sequências gênicas da região conservada 16S rDNA. As amostras foram coletadas de bovinos alimentados com duas dietas experimentais contendo diferentes relações volumoso e concentrado. Para amplificação da região ribossomal 16S rDNA foi feita PCR e a análise das sequências foi realizada pelos programas Phred/Phrap/Consed. As análises do BLAST permitiram identificar no tratamento com 70% de feno, 96 sequências relacionadas à família Methanobacteriaceae, 47 sequências a arqueias não cultiváveis e 60 sequências foram de arqueias desconhecidas e no tratamento com 30% de feno foram 125 sequências relacionadas à família Methanobacteriaceae, 42 sequências a arqueias não cultiváveis e 32 sequências foram de arqueias desconhecidas. A análise das sequências da região 16S rDNA de arqueias do rúmen bovino permitiu detectar maior número de sequências relacionadas com arqueias desconhecidas no tratamento com 70% de feno.
Palavras-chave: Análise molecular, metanogênicas, rúmen bovino, PCR.
INTRODUCTION
Methanogenic archaea inhabits a specific metabolic niche; they are strictly anaerobes and produces methane (CH4), an important energy source known as biogas. These organisms are found many in anaerobic environments, from deep aquatic environments like sea sediments to, the rumens and large intestines of herbivorous or used in biodigestors. The methanogenic organisms are present in the rumen in large quantities, ranging from 107 to 109 cells/mL of ruminal liquid, depending on the diet provided to the animal (Kamra, 2005). One of the largest sources of CH4 emission from agricultural activities is the enteric fermentation of ruminants (Olesen et al., 2006). The energetic loss due to ruminal methanogenesis causes economic prejudice against ruminants, since 3% to 13% of the gross energy intake of these animals can be lost as methane. Modern society demands high-yield food production systems with low pollutant emissions. Based on it, low-yield producing animals are major collaborators to the annual CO2 and CH4 gases production. One objective of ruminal microbial scientific research has been to develop methods to redirect the energetic losses due to CH4 into specialized products such as milk and meat, promoting better nutrient conversion (Nagajara, 2003).
Broader knowledge about microbial diversity allows a better understanding of the role of microorganisms in the environment and increases awareness of their interactions with other biodiversity components (Hunter-Cevera, 1998). Due to the high microbial diversity represented by the non-cultured organisms and the limitations of cultivation of extremophilic organisms (e.g., hyperthermophiles, psychrophiles, and others) in laboratory conditions, it is essential that new technologies be adopted to more broadly explore microbial diversity. Molecular techniques based on metagenomic approaches use total genomic DNA (gDNA) directly extracted from the environment, and, through PCR amplification, cloning and sequencing of the ribosomal DNA (rDNA) gene, make the identification of unknown microorganisms possible (Kent & Triplett, 2002).
The bacteria systematic committee has suggested the adoption of techniques that use 16S rRNA to identify, and even to classify, bacterial strains, (Stackebrandt et al., 2002). According to Hongoh et al. (2003), this advance has been extensively used in studies related to complex microbial communities found within the environment. The goal of this study was to detect the presence of archaea, through sequencing of the 16S rDNA conserved region, in the solid fraction sampled from the rumen of animals fed with experimental diets that had different roughage to concentrate ratios.
MATERIALS AND METHODS
Animal management
Two rumen-cannulated, crossbred animals, half Angus and half Nellore (AN), were placed into experimental stalls where each received one type of diet. The rougha-ge prepared and offered to the animals was Tifton 85 hay ground without siege. One animal received 70% hay and 30% concentrate (70H:30C, T1) and the second one received 30% hay and 70% concentrate (30H:70C, T2). The concentrate composition in the T2 diet consisted of 35.2% citric pulp, 19% soybean hulls, 15% sunflower seeds and 0.8% mineral supplementation. In the T1 diet, the concentrate content was 14.2% citric pulp, and 15% sunflower seeds and 0.8% mineral supplementation.
DNA extraction and PCR
The bacteria in the solid fraction from the rumen was performed according to Ezequiel et al. (2002). Next, genomic DNA was extracted following the methodology proposed by Hensiek et al. (1992). The primer sequences used in the PCR reactions to amplify the 16S rDNA were designed according to Whitford et al. (2001)After optimization of the PCR conditions, the final reactions were set up as follows: 200 ng of DNA, 1U Taq DNA polymerase in 1X buffer (Invitrogen), 6.0 mM magnesium chloride (MgCl2), 0.1 mM of each dNTP, 50 pmol of each primer, and sterile Milli-Q water to complete the final volume up to 20 mL (Whitford et al., 1998). The PCR amplifications were performed for 94ºC for 5 min followed by 35 cycles of 95ºC for 1 min, 51ºC for 2 min, and 72ºC for 3 min, then 72ºC for 10 min and 4ºC to cool down the samples (Hales et al., 1996). Amplicons were visualized by electrophoresis on 1.5% agarose gels ethidium bromide stained run at 75 V for 1.5 h.
Cloning and ligation reaction
PCR fragments amplified from the 16S rDNA were cloned into the pGEM-Teasy Vector, System I (Promega) following the manufacturers instructions. After bacterial transformation using E. coli DH10b cells, selected clones were grown in 150 mL of CircleGrow medium (Bio 101) containing ampicillin (100 mg/mL) for 22h at 37o C.
Plasmid DNA and sequencing
Stocked clones were cultured on Mega Titer plates containing 1 mL of CG medium plus 100 mg/ml ampicillin for 22 h at 37ºC. Plasmid DNA extraction was performed according to Sambrook & Russel, 2001. PCR reactions of the 16S rDNA region for sequencing were carried out as: 2 mL BigDye Terminator (Perkin Elmer), 5 pmol of primer, 5X buffer (400 mM Tris-HCl pH 9; 10 mM MgCl2) up to the final volume of 10 µl and 100-150 ng of DNA. For the sequencing PCR reaction, the universal SP6 primer was used (5 – ATT TAG GTG ACA CTA TAG -3). Plates were put into a thermocycler (MJ Research, Inv., model PTC-100) and then amplified as follows: 96ºC for 10 s, 52ºC for 5 s, and 60ºC for 4 min, repeated 34 times. Samples were stored at 4°C until use. Fragments were sequenced using an ABI 3100 capillary DNA sequencer. The sequences were later analyzed using Sequencing Analysis 3.4 software and the quality of the electropherograms were analyzed using Phred (Ewing et al., 1998), Phrap (Ewing & Green, 1998), Consed software (Gordon et al., 1998).
Phylogenetic analysis
Next, the sequences were analyzed online using the Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1997) for compare to other sequences in GenBank from the National Center for Biotechno-logy Information (NCBI). Sequence data of the inferred amino acid was aligned by use of CLUSTAL W ver.1.7 (Thompson et al., 1994) with reference to the sequences obtained from the GenBank using neighbour-joining method (Saitou & Nei, 1987). The tree was evaluated by using the bootstrap test based on 1000 resamplings (Felsenstein, 1985). The list of sequences obtained and deposited on the NCBI database can be found at http://lbmp.fcav.unesp.br/rumen, whichs the accession numbers are FJ 586890 to FJ 587072 and FJ 586726 to FJ 586889 for T1 and T2 diets, respectively.
The sequences were also compared to the Ribosomal Database (http://rdp.cme.msu.edu/index.jsp). The DOTUR software was used for statistical analysis (Schloss & Handelsman, 2005). To estimate the number of operational taxonomic units (OTUs) with this software, the 97%, 95% and 90% confidence intervals were considered. The distances among known and unknown sequences were calculated to compare the 16S rDNA sequences. Such comparisons yielded distances of approximately 3% for species, 5% for genera, and 10% for families.
The standard archaea lineages used in this research were: Halobacterium halobium (M11583), Methanobrevibacter smithii (AF054208), Methanobacterium bryantii (M59124), Methanosarcina mazeii (U20151), Methanococcoides burtonii (X65537), Metha-nolobus taylorii (U20154), Methanomicrobium mobile (M59142) and Methanosphaera stadtmanii (M59139) (Whitford et al., 2001).
RESULTS AND DISCUSSION
The cloning and sequencing techniques of the 16S rDNA were efficient in the phylogenetic evaluation of the ruminal archaea. For a preliminary identification of the clones, approximately 600 sequences were analyzed and then compared by the BLAST program on against the GenBank database (http://lbmp.fcav.unesp.br/rumen).
Based on the BLAST results, in the T1 diet, 96 sequences relating to the Methanobacteriaceae family, 47 sequences matching non-cultured archaea, and 60 sequences matching unknown archaea were found. In the T2 diet, 125 sequences relating to the Methanobacteriaceae family, 42 sequences matching non-cultured archaea, and 32 sequences matching unknown archaea were found. In total, 203 and 199 sequences were analyzed for each diet, respectively.
Statistical analysis using the DOTUR software allowed us to obtain relationships among the analyzed sequences at several taxonomic levels. In the T1 diet, we identified species for 25 sequences, genera for 23 sequences and families for 18 sequences. In the T2 diet, we identified species for 31 sequences, genera for 25 sequences and families for 21. Additionally, in the T1 diet, the sequences of 37 clones did not show any differences and in the T2 diet, over 100 sequences were similar.
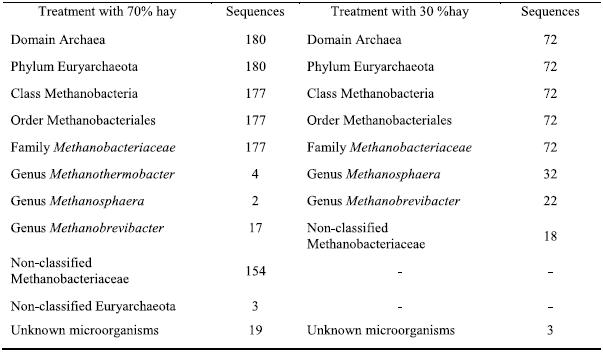
The sequences were also compared with sequences stored on the Ribosomal Database (RDP) (http://lbmp.fcav.unesp.br/rumen). We identified microorganisms of the Methanobacteriaceae family, including the Methanobrevibacter, Methanosphaera and Methanothermobacter genera. Table 1 shows the classification of the sequences analyzed in both diets based on the RDP classification. From the results in this table, it is clear that more sequences of non-classified archaea were found in the T1 diet, including some related to non-classified Euryarchaeota. Moreover, the genus Methanothermobacter was also present, whereas it was absent in the T2 diet. This result might be explained by the higher hay content of the diet; the large pieces of forage cause the fiber to pass through the digestive system more slowly, favoring the proliferation of cellulosic microorganisms in the rumen (Bryant & Burkey, 1953). The T2 diet resulted in a lower diversity since it returned fewer similar clones from different genera and it also comprised fewer unknown sequences.
Table 1 – The classification of the sequences in the two treatments with different proportions of hay, based on the analysis using the Ribosomal Database.
Ohene-Adjei et al. (2008) performed phylogenetic studies on the ruminal methanogenic archaea collected from sheep and used the Ribosomal Database to analyze the sequences. Furthermore, they also used the DOTUR software to analyze the DNA library. Similar to our study, they also found assorted sequences clones from the Metha-nobacteriacea family.
A similarity matrix of the aligned sequences was generated by the MEGA software, grouping potential OTUs (Figures 1 and 2). Figure 1 (representing the T1 diet) demonstrates that the majority of clone sequences were grouped along with the unknown methanogenic organisms, and few sequences were grouped with the standard species, Methanosphaera stadtmanii. Based on the phylogram that represents the T2 diet (Figure 2), some sequences were grouped next to Methanobrevibacter smithii and others along with the standard Methanos-phaera stadtmanii and another sequence grouped close to the Methanomicrobium mobile standard. While both unknown metagenomic species and the standard species, Methanosphaera stadtmanii, were found in the two treatments, the T2 diet had more clones grouped with Methanosphaera stadtmanii, indicating that more concentrated diets might select this species in the ruminal environment.
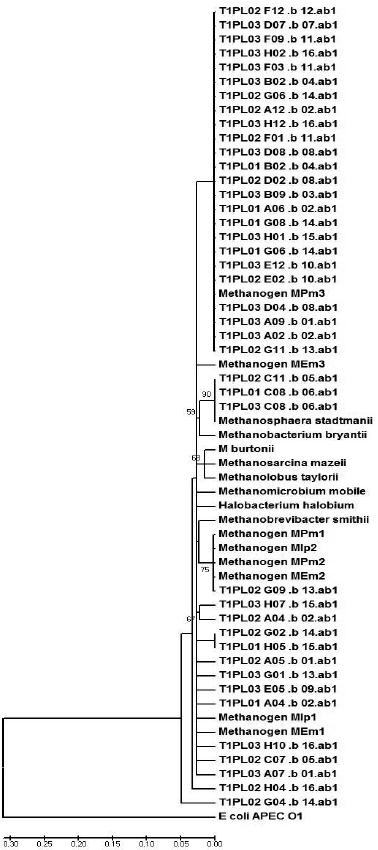
Figure 1 – The phylogram, grouped by phyla, of the partial sequences of the16S rDNA gene that were amplified from bovine that were fed 70% hay. The phylogram was generated using the Jukes-Cantor distance matrix by the neighbor-joining method. Bootstrap values for 1000 trees are shown at nodes; only values of ˃ 50% are shown. The scale represents the number of substitutions per base.
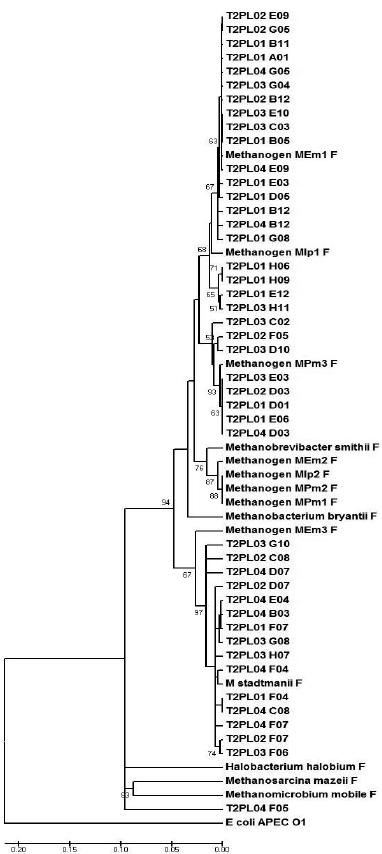
Figure 2 – The phylogram, grouped by phyla, of the partial sequences of the16S rDNA gene that were amplified from bovine that were fed 30% hay. The phylogram was generated using the Jukes-Cantor distance matrix by the neighbor-joining method. Bootstrap values for 1000 trees are shown at nodes; only values of ˃ 50% are shown. The scale represents the number of substitutions per base.
Due to archae implications with the global warming, several authors have been making efforts to detect and quantify these microorganisms in bovine rumen and other ruminants using for this purpose 16SrDNA or 16SrRNA in the phylogenetic determination of these methanogens. Pei et al. (2008) analyzed archaea diversity from the bovine rumen via 16S rRNA sequences, obtained OTU and carried out BLAST comparisons with sequences stored in GenBank. As in this work, their phylogenetic study identified clones related to microorganisms from the Methanobrevibacter, Methanosphaera and Methanomicrobium genera, additionally, they found unknown archaea and suggested that their results could indicate the existence of other methanogenic archaea in the rumen. Differently, they determined that some clones related to the Methanobacterium genera.
The present study determined in both types of diets sequences that related to Methanobacteriales class, Methanobacteriaceae family and Methanobrevibacter genera. Some studies showed that most methanogenic microorganisms detected in the rumen belong to the Methanobacteriaceae family and the species more frequently found are of the Methanobrevibacter genera (Skillman et al., 2004; Wright et al., 2004). Tajima et al. (2001) studied the molecular diversity of archaea found inside bovine rumen using the 16S rRNA. They used 2 pairs of primers to assemble the genetic library and verified many sequences from Methanomicrobium and Methanobrevibacter genera. Likewise, Skillman et al. (2006) amplified the 16S rRNA genes from archaea by PCR with specific primers, assembled the library and later sequenced the clones. They uncovered sequences related to many archaea species, such as Methanobrevibacter ruminantium, Methanobrevibacter smithii, Methanobrevibacter thaueri and Methanosphaera stadtmanii. The authors suggest that the diversity of methanogenic organisms recovered can be affected by the primers used in the experiment. Wright et al. (2007) studied the molecular diversity of bovine ruminal archaea coming from two distinct geographic locations in Canada. Among all the sequences analyzed, some matched M. ruminantium while some clones were unidentified. Other sequences were similar to the Methanobacteriales, Methanomicrobiales and Methanosarcinales orders, but no matches were found to the Methanobacterium, Methanomicrobium and Methanosarcina genera. The present study did not find any sequences closely related to these genera either. However, various species from these three genera have been identified previously (Tajima et al., 2001; Regensboge-nova et al., 2004; Shin et al., 2004).
In our study we assessed archaea present in the ruminal solid fraction and found that most clones grouped next to Methanobacteriaceae family. Shin et al. (2004), studied the phylogeny of archaea found in the fluid and solid fractions from the rumen and the ruminal epithelium of bovines by means of archaea specific primers. In the epithelium and fluid, they discovered a larger group of clones resembling the Methanomicrobiaceae family, while in the solid phase, the majority of clones were grouped next to the Methanobacteriaceae family
Several studies demonstrate that the majo-rity of microorganisms detected in the rumen belong to the Methanobacteriaceae family, and often the species most commonly found belong to the Methanobrevibacter genus (Skillman et al., 2004; Wright et al., 2004). This may be due to differences in the sample preparation, the animal diet or the geographic region sampled. Clearly, more studies are needed on the effects of diet and animal species on the diversity of methanogens in the rumen (Whitford et al., 2001).
CONCLUSIONS
In this study we found sequences of clones that grouped close to the standard Methanosphaera stadtmanii on diets with 70% of hay and 70% of concentrate, while, the diet with more concentrate allowed groupings with Methanobrevibacter smithii and Methanomicrobium mobile patterns. Through the analyses of the Ribosomal Database, the Methanosphaera, Methanobrevibacter and Methanothermobacter genera were determined, the latter being found only on the diet containing higher amount of roughage. The results lead to the conclusion that the type of diet given to animals, with higher or lower proportion of roughage interferes in the ruminal environment, once the diet containing higher proportion of hay was the one which showed larger number of sequences related to unknown methanogens and larger diversity of methalogenic archae in bovine rumen.
Acknowledgements
This study was funded by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
REFERENCES
Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. (1997) ─ Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25: 3389-3402.
Bryant, M.P. & Burkey, L.A. (1953) ─ Numbers and some predominant groups of bacteria in the rumen of cows fed different rations. Journal Dairy Science 36: 213-218.
Ewing, B. & Green, P. (1998) ─ Base-calling of automated sequencer traces using Phred II. Error probabilities. Genome Research 8: 186-194.
Ewing, B.; Hillier, L.; Wendl, M.C. & Green, P. (1998) ─ Base-calling of automated sequencer traces using Phred I. Accuracy assessment. Genome Research 8: 175-185.
Ezequiel, J.M.B., Melício, S.P.L.; Sancanari, J.B.D.; Ferreira, R.N.; Feitosa, J.F. (2002) ─ Quantificação das bactérias sólido-aderidas, bactérias e protozoários líquido-associados do rúmen de bovinos jovens alimentados com amiréia. Revista Brasileria de Zootecnia 31: 707-715.
Felsenstein, J. (1985) ─ Confidence limits on phylogenies: an approach using the boot strap. Evolution 39, 783-791.
Gordon, D.; Abajian, C. & Green, P. (1998) ─ Consed: A graphical tool for sequence finishing. Genome Research 8: 195-202.
Hales, B.A.; Edwards, C.; Ritchie, D.A.; Hall, G.; Pickup, R.W.; Saunders, J.R. (1996) ─ Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis. Applied and Environmental Microbiology 62: 668-675.
Hensiek, R.; Krupp, G. & Stackebrandt, E. (1992) ─ Development of diagnostic tract. Syst. Applied Microbiology 15: 123-128.
Hongoh, Y.; Ohkuma, M. & Kudo, T. (2003) ─ Molecular analysis of bacterial oligonucleotide probes for four lactobacillus species occurring in the intestinal microbiota in the gut of the termite Reticulitermes spe-ratus (Isoptera, Rhinotermitidae). FEMS Microbiology Ecology 44: 231–242.
Hunter-Cevera, J.C. (1998) ─ The value of microbial diversity. Current Opinion in Microbiology 1: 278-285.
Kamra, D.N. (2005) ─ Rumen microbial ecosystem. Current Science 89: 124-134.
Kent, A.D.; Triplett, E.W. (2002) ─ Microbial communities and their interactions in soil and rhizosphere ecosystems. Annual Review Microbiology 56: 211-236.
Nagaraja, T.G. (2003) ─ Response of the Gut and Microbial Populations to Feedstuffs: The ruminant story. In: Proc. 64th Minnesota Nutrition Conference. St. Paul, MN., pp.64-77.
Ohene-Adjei, S.; Chaves, A.V.; McAllister, T.A.; Benchaar, C.; Teather, R.M.; Forster, R.J. (2008) Microbial Eco-logy 56: 234-242.
Olesen, J.E.; Schelde, K.; Weiske, A.; Weisbjerg, B. M.R.; Asman, W.A.H.; Djurhuus, J. (2006) ─ Modelling greenhouse gas emissions from European conventional and organic dairy farms. Agriculture, Ecosystems and Environment 112: 207-220.
Pei, C.; Mao, S. & Zhu, W. (2008) ─ Molecular diversity of rumen Archaea from Jinnan cattle. Acta Microbiologica Sinica 48: 8-14. [ Links ]
Regensbogenova, M.; Mcewan, N.R.; (2004) ─ A re-appraisal of the diversity of the meyhanogens associated with the rumen ciliates. FEMS Microbiology 328:307-313.
Saitou, N.; Nei, M. (1987) ─ The neighbor-joining method: a newmethod for reconstructing phylogenetic trees. Molecular Biology and Evolution 4: 406-425.
Sambrook, J; Russel, D.W. (2001) ─ Molecular cloning - a laboratory manual. 3nd ed. Cold Spring Harbor Laboratory Press, New York.
Schloss, P.D.; Handelsman, J. (2005) ─ Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Applied Environment Microbiology 71: 1501-1506.
Shin, E.C.; Choi, B.R., .; Lim, W.J.; Hong, S.Y.; An, C.L.; Cho, K.M.; Kim Y.K.; An, J.m.; Kang, J.M.; Lee, S.S.; Kim, H.; Yun, H.D. (2004) ─ Division of Applied Life Science, Research Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence. Anaerobe 10: 313-319.
Skillman, L.C.; Evans, P.N. (2004) ─ 16S ribosomal DNA-directed PCR primers for ruminal methanogens and identification of methanogens colonising young lambs. Anaerobe 10: 277–285.
Skillman, L.C.; Evans, P.N., Strömpl, C.; Joblin, K.N. (2006) ─ 16S rDNA directed PCR primers and detection of methanogens in the bovine rumen. Letters Applied Microbiology 42: 222-228.
Stackebrandt, E. W.; Frederiksen, G. M.; Garrity, P. A.; Grimont, P.; Kampfer, M. C.; Maiden, X.; Nesme, R.; Rossello-Mora, J.; Swings, H. G.; Truper, L.; Vauterin, A. C.; Ward & Whitman, W. B. (2002) ─. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. International Journal of Systematic and Evolutionary Microbiology 52: 1043 -1047.
Tajima, K.; Nagamine, T., Matsui, H.; Nakamura, M.; Aminov, R.I. (2001) ─ Phylogenetic analysis of archaeal 16S rRNA libra-ries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiology Letters 1: 67-72.
Thompson, J.D.; Higgins, D.G. & Gibson, T.J. (1994) ─ CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research 22: 4673–4680.
Whitford, M.F.; Forster, R.J.; Beard, C.E.; Gong, J.; Teather, R.M. (1998) ─ Phylogenetic analysis of rumen bacteria by comparative sequence analysis of cloned 16S rRNA genes. Anaerobe 4: 153–163.
Whitford, M.F.; Teather, R.M. & Forster, R.J. (2001) ─ Phylogenetic analysis of methanogens from the bovine rumen. Microbio-logy 1: 5.
Wright, A.D.G.; Williams, A.J.; Winder, B.; Christophersen, C.T.; Rodgers, S.L.; Smith, K.D (2004) ─ Molecular diversity of rumen methanogens from sheep in Western Australia. Applied Environmental Microbiology 70: 1263–1270.
Wright, A.D.G.; Auckland, C.H. & Lynn, D.H. (2007) ─ Molecular diversity of me-thanogens in feedlot cattle from Ontario and Prince Edward Island, Canada. Applied Environmental Microbiology 200: 67-72.
Recepção/Reception: 2009.10.01
Aceitação/Acception: 2010.07.07

