










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.26 no.6 Porto dez. 2012
ARTIGO DE REVISÃO
Stresse, Catecolaminas e Risco Cardiovascular
Stress, catecholamines and cardiovascular risk
Mafalda Costa Pereira1, Laura Ribeiro2
1 Faculdade de Medicina da Universidade do Porto
2 Departamento de Bioquímica e Centro de Educação Médica, Faculdade de Medicina da Universidade do Porto
RESUMO
A doença cardiovascular (DCV) é a principal causa de mortalidade a nível mundial e prevê-se que o número de mortes aumente. Em determinados indivíduos, alguns factores de risco cardiovascular, como a resistência à insulina, obesidade, hipertensão arterial e dislipidemia, têm tendência a agregar-se numa entidade única, a denominada Síndrome Metabólica (SM). A prevalência dessa síndrome está a aumentar em todo o mundo, cada vez que mais pessoas adoptam o estilo de vida ocidental. Entre as principais características desse estilo de vida, além do balanço de energia positivo e dos comportamentos prejudiciais à saúde, destaca-se o stresse psicossocial. A resposta ao stresse leva à activação de dois importantes sistemas neurohumorais, o eixo hipotálamo-hipófise-suprarrenal e o sistema simpato-adreno-medular, que libertam cortisol e catecolaminas, respectivamente, exercendo uma acção crucial na função cardiovascular e no metabolismo energético. No entanto, esses sistemas, sob influência dos padrões de vida actual, nomeadamente quando repetidamente activados por longos períodos, podem deixar de ter uma acção adaptativa e conduzir a doença. Estudos recentes têm realçado o impacto do stresse psicossocial na DCV, sugerindo que o aumento da reactividade ao stresse, de forma sustentada, pode ser um factor preditivo da SM e eventos cardíacos adversos. Além disso, o aumento do tempo de recuperação após o evento stressor também se associa a risco cardiovascular. Estas evidências, podem ser úteis na prevenção e no tratamento da DCV. De facto, estudos recentes sugerem que intervenções a nível psicológico podem reduzir a recorrência e a mortalidade da DCV.
Palavras-chave: stresse psicossocial; catecolaminas; factores de risco cardiovascular; síndrome metabólica
ABSTRACT
Cardiovascular disease (CVD) is the leading cause of mortality worldwide and the number of deaths is expected to increase. in certain individuals, some cardiovascular risk factors such as insulin resistance, obesity, hypertension and dyslipidemia tend to cluster in a single entity, called Metabolic Syndrome (MS). the prevalence of this syndrome is increasing worldwide, as more people adopt the western lifestyle. Among the main features of this lifestyle, in addition to positive energy balance and adverse health behaviors, is psychosocial stress. the stress response leads to activation of two major neurohumoral systems, the hypothalamic-pituitary-adrenal axis and the sympathetic-adrenal medulla system which, through the release of cortisol and catecholamines respectively, exert crucial roles upon both cardiovascular function and energy metabolism. However, these systems under the influence of current living standards, particularly when activated repeatedly for prolonged periods, may fail to have an adaptive action. recent studies have highlighted the impact of psychosocial stress on cardiovascular disease, suggesting that increased reactivity to stress, in a sustained manner, can be a predictor of metabolic syndrome and adverse cardiac events. Furthermore, the increase in recovery time after the stressor event is also associated with cardiovascular risk. These evidences might be useful for both the prevention and treatment of CVD. in fact, recent studies suggest that psychological interventions reduce CVD recurrence and mortality.
Key-words: psychosocial stress; catecholamines; cardiovascular risk factors; metabolic syndrome
Introdução
A doença cardiovascular (DCV) é a principal causa de mortalidade a nível mundial. Estima-se que em 2004 17,1 milhões de pessoas morreram por DCV, representando 29% de todas as causas de morte, e que em 2030 aproximadamente 23,6 milhões de pessoas morrerão, principalmente por doença cardíaca e acidente vascular cerebral.1
Em alguns indivíduos, determinados factores de risco cardiovascular têm tendência a agregar-se. Uma dessas agregações é a designada Síndrome Metabólica (SM) em que há sobreposição entre resistência à insulina (RI), obesidade, hipertensão arterial (HTA) e dislipidemia.2
Embora a organização Mundial de Saúde (OMS) considere que a resistência à insulina seja um factor central da SM, estudos mais recentes (e de acordo com a Federação internacional de Diabetes) têm sugerido que a obesidade central/visceral é o componente mais importante e influente na patogénese dessa síndrome.3
A prevalência da SM é elevada (variando de 1050%) e a sua incidência continua a aumentar em todo o mundo, à medida que mais pessoas adoptam o estilo de vida ocidental.4-5 as principais características desse estilo de vida incluem um balanço de energia positivo (excesso de ingestão alimentar e baixa actividade física), baixa qualidade dos alimentos (densamente energéticos e pobres em micronutrientes), alteração dos ritmos biológicos e o stresse (psicossocial, prolongado e continuado no tempo).6
Apesar do balanço de energia positivo, e dos comportamentos prejudiciais à saúde, serem frequentemente considerados os principais responsáveis pelas proporções actuais da obesidade, e dos restantes componentes da SM, estudos mais recentes têm realçado o impacto dos factores psicossociais, como o nível de stresse psicológico, nestas patologias.7-9
O termo stresse tem vindo a ser definido como um estado de ameaça à homeostasia corporal, após a exposição a forças adversas intrínsecas ou extrínsecas (imprevisíveis e/ou incontroláveis10), denominadas stressores, que mobilizam um complexo espectro de respostas fisiológicas e comportamentais, com o objectivo de restabelecer essa mesma homeostasia.11
A resposta ao stresse leva à activação de dois importantes sistemas neurohumorais, o eixo hipotálamo hipófise-suprarrenal (HPA), em que o cortisol é o principalefector, eo sistema simpato-adreno-medular (SAM), que envolve a libertação das catecolaminas (CA), adrenalina (AD) e noradrenalina (NA), entre outros.11
A cascata neuroendócrina clássica é iniciada pela secreção central da NA e de outros mediadores químicos, os quais estimulam a libertação da hormona libertadora de corticotrofina (CRH) e vasopressina pelos neurónios dos núcleos paraventriculares do hipotálamo para o sistema portal hipofisário. A CRH induz o locus ceruleus ao nível da ponte e medula espinhal a segregar NA, que, consequentemente, leva à libertação de acetilcolina (ACH) pelas fibrassimpáticas dos nervos esplâncnicos, estimulando amedula da suprarrenal (MSR) a produzir AD, que, por sua vez, potencia o eixo HPA e a actividade do sistema nervoso simpático (SNS). Em poucos segundos, a CRH induz igualmente a secreção de corticotrofina (ACTH) para a circulação que vai estimular o córtex suprarrenal a produzir cortisol, o efector final do eixo HPA.12
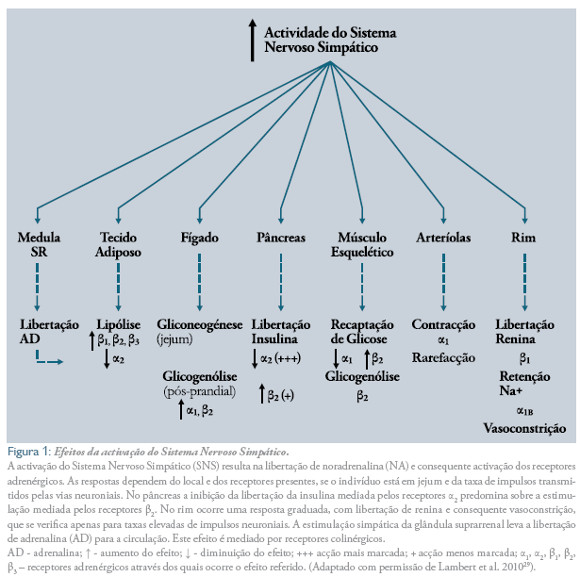
Walter Cannon (1932) descreveu que perante umaameaça o organismo responde através de uma activaçãodo sistema SAM, que o prepara para atacarou fugir (resposta fight or flight).6,13 Durante esta resposta, as funções cerebrais (do hipocampo,córtex pré- frontal e amígdala) e a atenção elevamse, bem como a frequência cardíaca (FC) e respiratória12. Por outro lado, os vasos sanguíneos contraem na maioria dos leitos vasculares e dilatam a nível muscular, redireccionando o fluxo sanguíneo para proporcionar melhor perfusão e desempenhodo cérebro, coração e músculos. De acordo com as necessidades energéticas, aumenta a mobilização de nutrientes, sobretudo glicose e lípidos, através da glicogenólise muscular, gliconeogénese e glicogenólise hepática e da lipólise nos adipócitos. Estes efeitos são ainda reforçados pelo estímulo da secreção de glicagina e inibição da secreção de insulina9,11,14(Figura 1 ). Todas as funções que não são necessárias à sobrevivência imediata ao stresse são diminuídas(apetite, função reprodutora, resposta à dor, função imunológica, etc.).11
Anos mais tarde, Hans Selye descreveu que a resposta ao stresse provoca os mesmos efeitos fisiológicos independentemente do estímulo aplicado, denominando-a de Síndrome de Adaptação Geral. Esta resposta desenvolve-se em três fases: fase de alarme (identificação do stressor), fase de resistência (tentativa de adaptação – coping) e uma fase de exaustão (os recursos são esgotados, havendo desregulação da resposta normal).15
No entanto, actualmente, o stresse é um conceito mais abrangente que engloba não só o estímulo mas também a resposta fisiológica, e que depende de um processo de avaliação individual (no qual o cérebro parece ser um órgão fundamental16), sendo modulado por múltiplos factores, incluindo características da personalidade que influenciam a noção de controlo da situação e o estilo de coping, factores sócio ambientais, nomeadamente o suporte psicológico, e características orgânicas do indivíduo. Nesse senti-do, cada indivíduo avalia um determinado stressor como um desafio (eustresse) ou ameaça (distresse).13,17 Chrousos (1992) introduziu pela primeira vez o conceito de desordens relacionadas com o stresse.18 Embora seja evidente que a activação destes sistemas teve um papel essencial para a sobrevivência humana e animal ao longo da evolução, sob influência dos padrões de vida actual, nomeadamente quando repetidamente activados por longos períodos, a sua acção pode deixar de ser adaptativa e, como tal, causar doença.6,9,19
Mais recentemente, os termos alostase e carga alostática foram propostos para explicar mais adequadamente a resposta fisiológica ao stresse e ultrapassar a ambiguidade desse conceito.10,20
Sterling e Eyer (1988)21 introduziram o termo alostase, isto é, a permanência de estabilidade após a mudança, através da acção de um sistema biológico dinâmico, que permite a adaptação a novas situações(físicas, psicológicas e ambientais), restabelecendo a homeostasia.13,20 Actualmente acredita-se que essa adaptação define um novo estado de equilíbrio, que não retorna ao nível prévio, e que, por isso, se a ameaça for contínua, pode ter efeitos prejudiciais para o organismo.22
Mc Ewen descreveu quatro situações de carga alostática: stressores frequentes ou contínuos; incapacidade de inactivação das respostas de alostase após cessação do evento stressor; resposta adaptativa inadequada, levando à activação de outros sistemas contra-reguladores, e ausência de habituação em resposta à exposição repetida dos mesmos estímulos.23
A carga alostática por si só não resulta em dano para o organismo e o stresse não é uma doença, mas quando se eleva a níveis em que os próprios mediadores da resposta alostática (insulina, cortisol, CA, etc.) começam a alterar a função normal, pode causar doença, designando-se nesse caso por sobrecarga alostática.13,16 A hiperestimulação dessa resposta pode ser entendida como stresse crónico.10
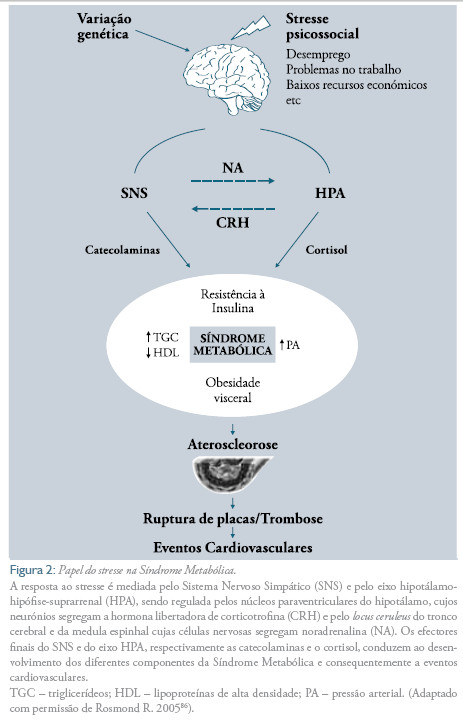
o stresse crónico pode assim ter efeitos prejudiciais à saúde. o cortisol e as CA são mediadores importantes desses efeitos ao longo do tempo, levando progressivamente, por exemplo, ao desenvolvimento de várias manifestações da SM e a eventos cardiovasculares11,19,24 (Figura 2).
Catecolaminas e suas acções
A AD e a NA são as principais CA envolvidas na regulação da resposta ao stresse, exercendo efeitos marcados na função cardiovascular, no metabolismo energético e em muitos outros processos fisiológicos.11-12 A NA é a principal CA libertada pelos neurónios pós-ganglionares do sistema simpático e a AD constitui a principal CA libertada pela MSR. A síntese das CA inicia-se no citoplasma dos nervos simpáticos, ou de células cromafins, pela hidroxilação do carbono 3 de uma molécula de tirosina, através da acção da hidroxílase da tirosina (passo limitante), para formar dihidroxifenilalanina (DOPA). Esta, por sua vez, é descarboxilada para formar dopamina (DA),pela descarboxílasedosl-aminoácidosaromáticos, sendo depois transportada para vesículas secretoras, onde é posteriormente β-hidroxilada pela β-hidroxílase da dopamina (DßH) para dar origem à NA. A NA em grande parte é N-metilada pela N-metiltransférase da feniletanolamina (PNMT), uma enzima citoplasmática presente sobretudo na MSR, dando origem a AD, que é transportada para as vesículas, onde é armazenada juntamente com a NA.
A libertação de CA nas terminações nervosas simpáticas ocorre pela acção da ACH, oriunda de neurónios pré-ganglionares. Nas células cromafins da MSr a ACH liga-se a receptores nicotínicos ou muscarínicos e permite o aumento da concentração intracelular de cálcio (através do seu influxo por canais dependentes da voltagem e da libertação a partir de reservas intracelulares), promovendo a fusão das membranas das vesículas com a membrana celular e subsequente exocitose das CA.
as reservas de CA são repostas, além da síntese de novo, por transporte activo, através do transportador da NA (NET), que remove a maioria das CA previamente libertadas. o restante é captado pelo transportador extraneuronial (ENT) ou sofre difusão para a circulação. A maioria das CA recaptadas é sequestrada nas vesículas de armazenamento pelo transportador vesicular de monoaminas (VMAT).14
As duas principais enzimas envolvidas na degradação das CA são a monoaminoxídase (MAO) e a catecol-o-metiltransférase (COMT).25
A ADENA exercem os seus efeitos fisiológicos através da ligação a receptores membranares específicos. os receptores adrenérgicos são classificados em três grupos -α1, α2e β– que, por sua vez, se subdividem em α1A,B,D, α2 A,B,C e β1,2,3, respectivamente. Estes receptores estão acoplados a proteínas G.26
A nível vascular, as CA provocam contracção do músculo liso, principalmente via receptores α1 mas também a2 e vasodilatação via receptores β2. A nível cardíaco aumentam a FC e a força de contracção, aumentando o volume e a fracção de ejecção e finalmente o débito cardíaco (DC), sobretudo via receptores β1, mas também β2e α1. Além disso, as CA têm importantes efeitos a nível metabólico. Estas aminas, via receptores α1e β2 aumentam a glicogenólise e gliconeogénese hepática. No músculo esquelético, as CA promovem a glicogenólise via receptores β1, diminuem a recaptação de glicose via receptores α1 e aumentam-na via receptores β2. A lipólise, e consequente libertação de ácidos gordos livres (AGL), aumenta no tecido adiposo via receptores β1,2,3 e diminui via receptores α2. No pâncreas, as CA estimulam a produção de glicagina, e alteram a secreção de insulina, diminuindo-a ou aumentando-a via receptores α2e β2, respectivamente. No rim aumentam a secreção de renina via receptores β1e aumentam a vasoconstrição e a retenção de sódio, via receptores α1 (Figura 1 ).
Por outro lado, a libertação das CA, e a amplitude das suas acções, pode ser regulada por feedback negativo e positivo através de receptores pré-sinápticos, α2e β2, respectivamente.
De forma geral, a AD tem maior afinidade para os receptores β e a NA para os receptores a, por ex., relativamente aos receptores β2 a afinidade da AD é 10-50 vezes superior à da NA. Contudo,a afinidade da AD para os receptores a1 é superior na maioria dos órgãos e a NA tem maior afinidade (cerca de 10 vezes mais) para os β3 do que a AD. A afinidade para os receptores β1 é idêntica para as duas CA.14
Stresse e síndrome metabólica
Vários estudos nas últimas duas décadas demonstraram de forma inequívoca que os principais componentes da SM se associam a uma hiperactivação do sistema simpático.9,19,27 Além disso, várias evidências sugerem que a activação do sistema SAM, em resposta ao stresse psicossocial, pode predispor ao desenvolvimento de várias componentes que constituem essa síndrome9,28-29 (Figura 2).
Obesidade
Alguns autores sugerem que a obesidade visceral é o principal preditor da ocorrência da SM, mesmo quando comparada com a RI.3
Embora actualmente se saiba que o SNS está cronicamente activado nos indivíduos obesos30 (ao contrário do que propuseram Bray et al. em 198931), ainda está por esclarecer se a activação simpática é causa ou consequência da obesidade.29,32 Segundo Julius et al. (2000) o aumento da actividade simpática predispõe ao aumento do peso.33 por outro lado, outros autores34-36 defendem que a obesidade é uma das causas de hiperactivação do SNS. Curiosamente, vários estudos demonstram que na obesidade a actividade do SNS está aumentada mas a actividade da MSR está diminuída. Além disso, indivíduos que apresentam mais componentes da SM excretam níveis progressivamente mais elevados de NA na urina, mas mais baixos de AD.27, 29 De facto, na SM, a AD e NA parecem comportar-se de maneira diferente: enquanto que os níveis de NA se relacionam de forma positiva com a obesidade e o risco cardiovascular, a AD parece associar-se de forma inversa.27,32,37 os mecanismos que controlam a diferente libertação das duas CA na obesidade são ainda mal conhecidos.6
Um estudo em indivíduos caucasianos, após 18 anos de seguimento, mostrou que os níveis de AD associados ao stresse mental se relacionam negativamente com o índice de massa corporal (IMC) e o perímetro abdominal.32 por outro lado, esse estudo demonstrou que os níveis de NA se relacionam positivamente, embora de forma menos marcada, com o IMC.32 A diminuição da actividade da MSR e dos níveis de AD podem ter assim um papel preditor importante no aumento do peso corporal. Isto verifica-se mesmo após correcção para possíveis confundidores, como o nível de exercício físico, uma vez que a actividade da MSR aumenta com a melhor performance física e os indivíduos obesos são geralmente mais sedentários.27,32
A gordura corporal parece relacionar-se com os níveis de AD associados ao stresse e não com os níveis em repouso, sugerindo que a predisposição individual para responder ao stresse mental é um determinante importante no aumento do peso corporal.32 Além disso, uma quantidade crescente de evidências sugere que os receptores β-adrenérgicos têm um papel importante no desenvolvimento da obesidade.32 Ratinhos knockout para os receptores β1,2,3 desenvolvem obesidade maciça com dieta rica em gordura, comparativamente aos controlos.38 Por outro lado, polimorfismos dos receptores adrenérgicos β2e β3 em humanos associam-se a um aumento de peso.32,39-40 Em resposta ao stresse, a libertação de níveis baixos de AD (com afinidade sobretudo para os receptores β, activados em resposta ao stresse41), também pode levar ao aumento de peso.32 De acordo com isto, os índios pima têm uma das mais altas prevalências de obesidade em todo o mundo e têm menor actividade simpática a nível muscular42 e menor sensibilidade cronotrópica à activação β adrenérgica43 do que os indivíduos caucasianos.
Dislipidemia
Os mecanismos patofisiológicos através dos quais o sistema SAM pode contribuir para o desenvolvimento da dislipidemia na SM estão bem estabelecidos. Nos humanos, as CA regulam a lipólise através do balanço entre o efeito lipolítico dos receptores adrenérgicos β e do efeito antilipolítico dos receptores adrenérgicos a229 (Figura 1 ). o aumento dos níveis circulantes de CA induz a libertação de AGL a partir do tecido adiposo, que, por sua vez, servem de substrato para a síntese de novo de triglicerídeos e produção hepática de lipoproteínas de muito baixa densidade (VLDL).9,28,41 por outro lado, evidências revelam que os níveis de colesterol total (CT) e das suas fracções aumentam em resposta ao stresse psicossocial.32,41,44-45 isto reforça a ideia que um melhor perfil cardiovascular em parte pode ser explicado pela manutenção de níveis mais baixos de CA.45-46
Um estudo demonstrou que o aumento da NA em resposta ao stresse se associa de forma significativa com aumento do CT e de lipoproteínas de baixa densidade (LDL), independentemente da idade, do IMC e da pressão arterial média.45 Contudo, a AD parece estar associada a um perfil lipídico mais benéfico. Reaven et al. postularam que a baixa actividade da MSR e dos níveis de AD contribuem para anormalidades lipídicas; por sua vez, níveis elevados de AD relacionam-se negativamente com os níveis de triglicerídeos e positivamente com os níveis de lipoproteínas de alta densidade (HDl), independentemente do IMC, níveis de insulina e actividade física.47 De acordo com isto, o bloqueio dos receptores adrenégicos ß(para os quais a AD tem maior afinidade) diminui os níveis de HDL e aumenta os níveis de triglicerídeos.27
Resistência à insulina
A ligação entre activação do SNS, hiperinsulinemia e RI é complexa. Por um lado, verifica-se que níveis elevados de insulina podem promover a activação do SNS;34-35 por outro, o SNS pode predispor a RI,33,48 havendo um reforço recíproco.41 Deste modo, ainda se desconhece se a RI é causa ou efeito da activação do SNS, apesar de alguns autores já terem proposto alguns mecanismos através dos quais ocorre esta ligação.29-30
Está bem estabelecido que as CA aumentam a glicogenólise e a gliconeogénese hepática e diminuem a sensibilidade à insulina e a recaptação de glicose pelo músculo esquelético, conduzindo consequentemente à diminuição da tolerância à glicose e a RI. Por outro lado, as CA inibem a secreção de insulina e estimulam a de glicagina, o que também contribui para o aumento dos níveis de glicose no sangue.14 Além disso, em adipócitos isolados a estimulação adrenérgica ßinduz dessensibilização dos receptores da insulina.49 De acordo comisso, alguns estudos revelam que os indivíduos mais reactivos ao stresse do dia-a-dia têm aumento da libertação de CA e, consequentemente, níveis mais elevados de glicose em circulação.46 Curiosamente, reaven et al. postularam que os níveis de AD se associam negativamente com a RI.47
A actividade do SNS, por outro lado, através da libertação de NA causa vasoconstrição dos capilares e diminui o fluxo sanguíneo, dificultando a captação de glicose pelo músculo esquelético, local de maior RI.50-52 Confirmando esta hipótese, um estudo em indivíduos caucasianos revelou que a activação dos receptores adrenérgicos a pela NA, em resposta a um teste de stresse induzido pelo frio, se associa positivamente com os níveis de glicose em jejum e com a RI, após 18 anos de seguimento. Por sua vez, não se verificaram associações significativas com os níveis das CA plasmáticas em repouso ou em resposta a um teste de stresse mental (por activação β).41 Outro estudo longitudinal, realizado em indivíduos japoneses, demonstrou, após 10 anos de seguimento, que o aumento da actividade simpática é um factor preditivo de hiperinsulinemia.53 Estes achados sugerem que a hiperactivação simpática pode ser um factor predisponente para RI.41
Além dos mecanismos referidos, os AGL, libertados em resposta à acção das CA, podem competir com a glicose para a captação e metabolismo celular no músculo esquelético, podendo potenciar a redução da sensibilidade à insulina.30,54
Hipertensão arterial
Dado o papel central do sistema SAM na regulação da pressão arterial (PA), tem sido sugerido que a hiperestimulação simpática seja um factor etiológico para a HTA.19,55-56
A activação do sistema renina-angiotensina-aldosterona (SRAA) pelas CA aumenta a renina circulante, levando à formação de angiotensina II (AgII) e aldosterona. Estas hormonas, em conjunto com a vasopressina, aumentam o volume sanguíneo e consequentemente a pressão venosa central e o DC, através de efeitos renais como o aumento da vaso constrição (e consequente diminuição da taxa de filtração glomerular (TFG)) e da reabsorção de sódio e água.30 A AgII, além de provocar vasoconstrição, pode promover remodelação cardiovascular, levando a hipertrofia e apoptose celular.56 o aumento das CA circulantes reforça ainda esses efeitos na PA, através da sua acção no coração e nos vasos sanguíneos. No coração, as CA, sobretudo a AD,57 através dos receptores β1, exercem uma acção estimulante directa (inotropismo, cronotropismo, dromotropismo ebatmotropismo positivos), aumentando o DC e consequentementea PA. os receptores a1, embora presentes em menor densidade, têm um papel importante no aumento da sensibilidade dos miofilamentos ao cálcio. Por outro lado, níveis elevados de CA circulantes promovem vasoconstrição, especial-mente nos vasos de resistência pré-capilares (da pele, mucosa e rins) juntamente com marcada constrição venosa, mediada pelos receptores a (sobretudo a1). Consequentemente, a resistência vascular periférica (RVP) aumenta, o que contribui para o aumento da PA. A nível renal, o aumento da resistência vascular traduz-se ainda em diminuição da TFG e da excreção de sódio, potássio e cloro, contribuindo adicionalmente para o aumento da PA.14
Vários estudos revelam que em resposta a testes de stresse mental, há uma hiperactivação do SNS, com aumento da libertação de CA e aumento dos níveis de pressão arterial sistólica (PAS), pressão arterial diastólica (pAD) e FC. Nos indivíduos mais reactivos, as respostas cardiovasculares são mais intensas e o retorno da PAD aos níveis basais parece ser mais lento, quando comparado com os indivíduos menos reactivos.19,45-46 Além disso, os indivíduos que apresentam maior reactividade ao stresse têm níveis de PA mais elevados em repouso, que se associam proporcionalmente a aumentos da actividade da MSR e do SNS e consequentemente dos níveis de AD e NA, em comparação com os indivíduos menos reactivos que apresentam valores normais ou mesmo baixos de PA.27,45-46,58 Mais interessante é que num estudo realizado em jovens saudáveis, que foram seguidos durante 18 anos, ficou demonstrada uma associação entre hiperreactividade cardiovascular ao stresse e o desenvolvimento de HTA.44
Uma das explicações do papel do SNS no desenvolvimento de HTA foi avançada já em 1981 por rand e Majewski, sendo designada por Hipótese da AD. Segundo esta hipótese, o aumento da AD na fenda sináptica activa os receptores adrenérgicos ß2 pré-juncionais e desencadeia o aumento da libertação de NA pelos terminais simpáticos, o que pode representar uma etapa precoce no desenvolvimento de vasoconstrição sustentada e HTA.59 De facto, experiências com ratos espontaneamente hipertensos demonstraram que a remoção da MSR atenuou o desenvolvimento de HTA. Além disso, o efeito próhipertensivo da AD foi abolido através do tratamento com antagonistas β2.60 Contudo, a atenuação da HTA só ocorreu com a remoção da MSr numa fase precoce da vida, indicando que pode existir um período crítico de sensibilização dos receptores ß2 pré-sinápticos.61 Em humanos, a elevação dos níveis de AD plasmática, libertados pela estimulação repetida da MSR em resposta ao stresse, pode facilitar a libertação de NA, levando à activação cardiovascular e possivelmente ao desenvolvimento de HTA.26,62-64 Alguns estudos demonstram a libertação de NA em níveis proporcionais aos de AD na hipertensão e também em resposta ao stresse, corroborando o papel estimulante da AD, de acordo com a Hipótese da AD. 27,64
Em 1982, Björn Folkow propôs que a HTA estaria relacionada com um aumento anormal da resistência ao fluxo sanguíneo secundário a alterações da estrutura dos vasos da microcirculação, não sendo necessário que o tónus vascular estivesse aumentado.65
Por um lado, a activação repetida ou exagerada do SNS em resposta ao stresse, pode causar aumentos intermitentes da PA, levando finalmente a remodelagem da estrutura microvascular, crescimento da túnica média dos grandes vasos e consequentemente a HTA sustentada e irreversível.44,55-56 Contudo, estudos realizados em cães, na tentativa de reproduzir aumentos sustentados e irreversíveis da PA como consequência de aumentos transitórios da PA, não foram bem sucedidos.44 por outro lado, embora a remodelação vascular possa ser dependente dos níveis de PA, a estimulação simpática pode ser, por si só, um factor trófico para a hipertrofia vascular, independentemente da PA. Assim os indivíduos hiperreactivos ao stresse com episódios frequentes de aumento da actividade simpática podem desenvolver aumentos sustentados da RVP total e consequentemente HTA.44,56 De acordo com isso, num estudo recente os níveis de AD e NA atingidos durante um teste de stresse psicológico, permitiram explicar 12,7% da variação da PAS após 18 anos de seguimento. Este estudo reforça a hipótese de que a actividade do SNS e concretamente a libertação de CA pode ter implicações no desenvolvimento de HTA, independentemente dos níveis de PA.44 No entanto, não há estudos conclusivos que suportem que a activação simpática tenha um papel independente napatogénese da remodelação vascular.Além disso, os bloqueadores ß não parecem influenciar a estrutura vascular nos indivíduos hipertensos.66-67 por sua vez, os antagonistas dos receptores da aldosterona, e os inibidores da enzima de conversão da angiotensina, parecem diminuir significativamente a resistência vascular.67-68 Assim, o efeito vasodilatador de alguns fármacos, especialmente os que interferem com o SRAA, parece influenciar a estrutura vascular.66
Considerando todos os mecanismos acima mencionados, entende-se facilmente como o stresse crónico, caracterizado por uma hiperactivação do sistema SAM e libertação maciça de CA, se relaciona com o aumento da incidência de SM. Vários estudos prospectivos confirmam que o stresse crónico pode ser um factor preditivo dessa síndrome.28,69
Stresse e função cardíaca
A hiperactivação do sistema SAM de forma sustentada em reposta ao stresse70 parece ter importantes efeitos a nível cardíaco.19,57 Dependendo da predisposição individual, o stresse pode levar ao desenvolvimento de doença cardíaca isquémica, enfarte agudo do miocárdio, insuficiência cardíaca, arritmias e em última instância à morte súbita.71
Um dos mecanismos subjacente aos efeitos das CA no coração parece ser dependente do aumento de cálcio nos cardiomiócitos, sobretudo através da activação dos receptores β1 acoplados à proteína G estimulatória (Gs), que leva à estimulação da adenilcíclase (AC), aumento de adenosina monofosfato cíclico (AMpc) e finalmente à activação da fosfocínase A (PKA).72
As CA, sobretudo a AD, aumentam a frequência e a força de contracção do coração (via receptores β1 e a1) e consequentemente o trabalho cardíaco,o que aumenta as necessidades de oxigénio, podendo levar a hipóxia. Por outro lado, sobretudo a NA, através dos receptores α1, pode provocar espasmo das artérias coronárias, diminuição do fluxo sanguíneo e da perfusão do miocárdio e consequentemente do fornecimento de oxigénio, podendo levar a isquemia. Além disso, o consumo das reservas de energia do miocárdio, sob a forma de adenosina trifosfato (ATP) (pelo aumento da actividade cardíaca em resposta às CA), pode levar a alterações bioquímicas (alteração do metabolismo lipídico e da glicose e consequente depleção de glicogénio e deposição lipídica no tecido cardíaco), alterações eléctricas (disfunção de proteínas transportadoras), aumento da permeabilidade membranar e alterações estruturais (edema e inflamação intersticial, ruptura e necrose de fibras miocárdicas, tumefacção das mitocôndrias, hemorragia subendocárdica, entre outras), que podem causar danos celulares irreversíveis.57,73
Outro aspecto importante do efeito das CA a nível cardíaco é a diminuição dos níveis de magnésio, um importante activador de enzimas envolvidas na transferência de cálcio74 e de fosfato75 para a mitocôndria para produzir ATP. Além disso, através da oxidação das CA, pode haver, por um lado desgaste de oxigénio76 e consequentemente desacoplamento da fosforilação oxidativa e insuficiência coronária anóxica e, por outro, produção de espécies reactivas de oxigénio, que se associam a alterações membranares no sarcolema, retículo sarcoplasmático (RS) emitocôndrias. A diminuição da actividade do trocador sódio-cálcio no sarcolema e da bomba de cálcio no sarcolema e RS contribuem para o aumento do cálcio intracelular. O stresse oxidativo pode também causar danos directos na membrana dos cardiomiócitos, havendo perda do conteúdo celular. as alterações associadas à sobrecarga de cálcio e à diminuição dos níveis de ATP associam-se naturalmente a disfunção da contractilidade cardíaca. Além dos efeitos referidos, o aumento do cálcio intracelular, pode levar por si só a arritmias.57 Confirmando estes mecanismos, verifica-se que o bloqueio dos canais de cálcio parece prevenir lesões de necrose do miocárdio em resposta à administração de CA.77
Por outro lado, os receptores a, através da activação das cínases de proteínas activadas por mitogénios (MAPK),14 podem levar a proliferação, hipertrofia e migração de células musculares lisas e de fibroblastos, levando consequentemente à remodelação do tecido cardíaco.78 os receptores β, através da produção de AMpc79 e de factores de crescimento que actuam de forma autócrina,80 podem ter efeitos semelhantes. Além disso, através do aumento do cálcio, a estimulação dos receptores β1 activa a cínase de proteínas II dependente de cálcio/calmodulina (CaMKII),81 levando à apoptose das células cardíacas. Pelo contrário, os receptores ß2, acoplados à proteína G inibitória (Gi), podem, por um lado proteger o miocárdio da apoptose, inibindo o sistema AC-AMpc-pkA,82 e, por outro, promover a sobrevivência celular,22 por activação da cínase 3 de fosfoinositídeos (PI3K), que estimula a fosfocínase b (PKB).
Alguns estudos sugerem que os receptores β3, via proteína Gi, ou por activação da síntase do oxido nítrico endotelial, ambos conduzindo a uma diminuição do cálcio, podem contrabalançar os efeitos pró-arritmíco, cronotrópico, inotrópico e dromotrópico positivos dos receptores ß1, estando envolvidos numa resposta adaptativa que protege o miocárdio dos efeitos prejudiciais da activação do SNS, durante condições extremas de stresse. Contudo esta hipótese necessita de uma investigação mais vasta e aprofundada.57,83
Recentemente, uma metanálise que incluiu estudos prospectivos publicados entre 1986 e 2009 quantificou a associação entre a reactividade a testes laboratoriais de stresse mental e o consequente risco cardiovascular.84 Esta metanálise demonstrou que a hiperrreactividade ao stresse se associa a efeitos cardíacos adversos, incluindo aumento dos níveis de PAS/PAD e HTA. as associações referidas foram mais pronunciadas no sexo masculino, em indivíduos mais jovens (=18 anos) e naqueles seguidos por longos períodos de tempo (=3 anos). Além do aumento da reactividade ao stresse, o aumento do tempo de recuperação após o evento stressor (sobretudo pela elevação sustentado da PAS e FC), também se associa a risco cardiovascular, nomeadamente com o aumento da espessura da íntima das artérias carótidas, além do aumento da PAS/PAD. Entre os diferentes tipos de stressores (tarefas cognitivas, entrevistas, falar em público, indução de emoções ou tarefas combinadas), apenas as tarefas que envolvem a cognição (testes aritméticos) se associaram significativamente a um maior risco cardiovascular.84 por outro lado, outra metanálise recente confirmou que intervenções a nível psicológico (através de técnicas de relaxamento, terapias cognitivo-comportamentais e alteração do ambiente social) podem ser úteis na presença de DCV.85 Esta metanálise mostrou que essas intervenções se associam a redução da FC, do CT, melhoria da depressão e do distresse, da qualidade de vida e da percepção de suporte, reduzindo dessa forma a recorrência de DCV e a mortalidade nesses indivíduos.85
Conclusão
Os mecanismos através dos quais o stresse crónico pode conduzir ao desenvolvimento de SM e consequentemente a um maior risco cardiovascular são vários. Nesse contexto, o número crescente de evidências do papel preditivo do stresse na DCV pode ser útil tanto na prevenção como no tratamento dessa patologia. Por isso, é importante que na prática clínica os médicos tenham em consideração os níveis de stresse, não só em doentes em risco, mas também nos indivíduos com doença cardíaca, e uma das formas de o fazer é incluir questões relacionadas com os níveis de stresse nas suas histórias clínicas.
Referências
1. World Health Organization. Fact Sheet on Cardiovascular Diseases. Geneva: Who; 2011 [Citado em 2011 30 Março] Available From: http://www.who.int/mediacentre/Factsheets/Fs317/En/Index.Html. [ Links ]
2. Stern MP, Haffner SM. Body Fat Distribution and Hyperinsulinemia as Risk Factors for Diabetes and Cardiovascular Disease. Arteriosclerosis 1986;6:123-30. [ Links ]
3. Tong J, Boyko EJ, Utzschneider KM, et al. Intra-Abdominal Fat Accumulation Predicts the Development of the Metabolic Syndrome in Non-Diabetic Japanese-Americans. Diabetologia 2007;50:1156-60. [ Links ]
4. Carnethon MR, Loria CM, Hill JO, Sidney S, Savage PJ, Liu K. Risk Factors for the Metabolic Syndrome: the Coronary Artery Risk Development in Young Adults (Cardia) Study, 1985-2001. Diabetes Care 2004;27:2707-15. [ Links ]
5. Wilsgaard T, Jacobsen BK. Lifestyle Factors and Incident Metabolic Syndrome. The Tromso Study 1979-2001. Diabetes Res Clin Pract 2007;78:217-24. [ Links ]
6. Azevedo A, Santos AC, Ribeiro L, Azevedo I. The metabolic syndrome. In: Soares R, Editor. Oxidative Stress, Inflammation and Angiogenesis in the Metabolic Syndrome. New York: Springer; 2009. P. 1-19. [ Links ]
7. Keith SW, Redden Dt, Katzmarzyk Pt, et al. Putative Contributors To the Secular Increase in Obesity: Exploring the Roads Less Traveled. Int J Obes (Lond) 2006;30:1585-94. [ Links ]
8. Shoelson SE, Herrero L, Naaz A. Obesity, Inflammation, and Insulin Resistance. Gastroenterology 2007;132:2169-80. [ Links ]
9. Vaccarino V, Bremner JD. Stresse Response and Metabolic Syndrome. The Hospital Physician Board Review Manual: Cardiology 2005;11:1-12. [ Links ]
10. Romero LM, Dickens MJ, CYR NE. The Reactive Scope Model -A New Model Integrating Homeostasis, Allostasis, and Stress. Horm Behav 2009;55:375-89. [ Links ]
11. Kyrou I, Tsigos C. Stress Hormones: Physiological Stress and Regulation of Metabolism. Curr Opin Pharmacol 2009;9:787-93. [ Links ]
12. Innes KE,Vincent HK,Taylor AG.Chronic Stress and Insulin Resistance- Related indices of cardiovascular disease risk, Part I: Neurophysiological responses and pathological sequelae. Altern Ther Health Med 2007;13:46-52. [ Links ]
13. Mcewen BS, Wingfield JC. What Is in A Name? Integrating Homeostasis, Allostasis and Stress. Horm Behav 2010;57:105-11. [ Links ]
14. Brunton LL, Lazo JS, Parker Kl. Goodman and Gilman -the Pharmacological Basis of Therapeutics. 11 ed. New York: Mcgraw-Hill; 2006. P. 158-170; 237-50. [ Links ]
15. Selye H. A Syndrome Produced By Diverse Nocuous Agents. 1936. J Neuropsychiatry Clin Neurosci 1998;10:230-1. [ Links ]
16. Mcewen BS, Gianaros PJ. Central Role of the Brain in Stress and Adaptation: Links To Socioeconomic Status, Health, and Disease. Ann N Y Acad Sci 2010;1186:190-222. [ Links ]
17. Leblanc VR. The Effects of Acute Stress on Performance: Implications for Health Professions Education. Acad Med 2009;84:S25-33. [ Links ]
18. Chrousos GP, Gold PW. The Concepts of Stress and Stress System Disorders. Overview of Physical and Behavioral Homeostasis. JAMA 1992;267:1244-52. [ Links ]
19. Brotman DJ, Golden SH, Wittstein IS. The Cardiovascular Toll of Stress. Lancet 2007;370:1089-100. [ Links ]
20. Logan JG, Barksdale DJ. Allostasis and Allostatic Load: Expanding the Discourse on Stress and Cardiovascular Disease. J Clin Nurs 2008;17:201-8. [ Links ]
21. Sterling P, Eyer J. Allostasis: A New Paradigm to Explain Arousal Pathology. In: Fisher S, Reason J, Editors. Handbook of Life Stress, Cogintion and Health. New York: J. Wiley & Sons; 1988. P. 629-49. [ Links ]
22. Santos IN, Spadari-Bratfisch RC. Stress and Cardiac Beta Adrenoceptors. Stress 2006;9:69-84. [ Links ]
23. Mcewen BS. Sex, Stress and the Hippocampus: Allostasis, Allostatic Load and the Aging Process. Neurobiol Aging 2002;23:921-39. [ Links ]
24. Figueredo VM. the Time Has Come For Physicians To Take Notice: the Impact Of Psychosocial Stressors on the Heart. Am J Med 2009;122:704-12. [ Links ]
25. Goldstein DS, Eisenhofer G, Kopin IJ. Clinical Catecholamine Neurochemistry: A legacy of Julius Axelrod. Cell Mol Neurobiol 2006;26:695-702. [ Links ]
26. Guimaraes S, Moura D. Vascular Adrenoceptors: An Update. Pharmacol Rev 2001;53:319-56. [ Links ]
27. Reims HM, Sevre K, Fossum E, Mellem H, Eide IK, Kjeldsen SE. Adrenaline during mental stress in relation to fitness, Metabolic risk factors and cardiovascular responses in young men. Blood Press 2005;14:217-26. [ Links ]
28. Brunner EJ, Chandola T, Marmot MG. Prospective effect of job strain on general and central obesity in the Whitehall Ii Study. Am J Epidemiol 2007;165:828-37. [ Links ]
29. Lambert GW, Straznicky NE, Lambert EA, Dixon JB, Schlaich MP. Sympathetic Nervous Activation in Obesity and the Metabolic Syndrome-Causes, Consequences and Therapeutic Implications. Pharmacol Ther 2010;126:159-72. [ Links ]
30. Chapman MJ, Sposito AC. Hypertension and Dyslipidaemia in Obesity and insulin resistance: Pathophysiology, Impact on Atherosclerotic Disease and Pharmacotherapy. Pharmacol Ther 2008;117:354-73. [ Links ]
31. Bray GA, York DA, Fisler JS. Experimental Obesity: A homeostatic failure Due to defective nutrient Stimulation of the Sympathetic Nervous System. Vitam Horm 1989;45:1-125. [ Links ]
32. Flaa A, Sandvik L, Kjeldsen SE, Eide IK, Rostrup M. Does Sympathoadrenal Activity Predict Changes in Body Fat? An 18-Y Follow-Up Study. Am J Clin Nutr 2008;87:1596-601. [ Links ]
33. Julius S, Valentini M, Palatini P. Overweight and Hypertension : A 2-Way Street? Hypertension 2000;35:807-13. [ Links ]
34. Landsberg L. Diet, Obesity and Hypertension: An Hypothesis Involving Insulin, the Sympathetic Nervous System, and Adaptive Thermogenesis. Q J Med 1986;61:1081-90. [ Links ]
35. Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. 1988. Nutrition 1997;13:65. [ Links ]
36. Grundy SM. What Is the Contribution of obesity to the metabolic syndrome? Endocrinol Metab Clin North Am 2004;33:267-82. [ Links ]
37. Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension2003;42:474-80. [ Links ]
38. Bachman ES, Dhillon H, Zhang CY, et al. Betaar signaling required for diet-induced thermogenesis and obesity resistance. Science 2002;297:843-5. [ Links ]
39. Masuo K, Katsuya T, Fu Y, Rakugi H, Ogihara T, Tuck ML. Beta2-and beta3-adrenergic receptor polymorphisms are related to the onset of weight gain and blood pressure elevation over 5 years. Circulation 2005;111:3429-34. [ Links ]
40. Masuo K, Katsuya T, Fu Y, Rakugi H, Ogihara T, Tuck ML. Beta2-adrenoceptor polymorphisms relate to insulin resistance and sympathetic overactivity as early markers of metabolic disease in nonobese, normotensive individuals. Am J Hypertens 2005;18:1009-14. [ Links ]
41. Flaa A, Aksnes TA, Kjeldsen SE, Eide I, Rostrup M. Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metabolism 2008;57:1422-7. [ Links ]
42. Spraul M, Ravussin E, Fontvieille AM, Rising R, Larson DE, Anderson EA. Reduced sympathetic nervous activity. A potential mechanism predisposing to body weight gain. J Clin Invest 1993;92:1730-5. [ Links ]
43. Tataranni PA, Christin L, Snitker S, Paolisso G, Ravussin E. Pima indian males have lower beta-adrenergic sensitivity than caucasian males. J Clin Endocrinol Metab 1998;83:1260-3. [ Links ]
44. Flaa A, Eide IK, Kjeldsen SE, Rostrup M. Sympathoadrenal Stress Reactivity Is a predictor of future blood pressure: An 18-year follow-up study. Hypertension 2008;52:336-41. [ Links ]
45. Wirtz PH, Ehlert U, Bartschi C, Redwine LS, Von Kanel R. Changes in plasma lipids with psycho social stress are related to hypertension status and the norepinephrine stress response. Metabolism 2009;58:30-7. [ Links ]
46. Flaa A, Mundal HH, Eide I, Kjeldsen S, Rostrup M. Sympathetic activity and cardiovascular risk factors in young men in the low, Normal, And high blood pressure ranges. Hypertension 2006;47:396-402. [ Links ]
47. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities-the role of insulin resistance and the sympathoadrenal system. N Engl J Med 1996;334:374-81. [ Links ]
48. Mancia G, Bousquet P, Elghozi JL, et al. The sympathetic nervous system and the metabolic syndrome. J Hypertens 2007;25:909-20. [ Links ]
49. Lonnroth P, Smith U. Beta-adrenergic dependent downregulation of insulin binding in rat adipocytes. Biochem Biophys Res Commun 1983;112:972-9. [ Links ]
50. Rocchini AP, Moorehead C, Katch V, Key J, Finta KM. Forearm resistance vessel abnormalities and insulin resistance in obese adolescents. Hypertension 1992;19:615-20. [ Links ]
51. Jamerson KA, Julius S, Gudbrandsson T, Andersson O, Brant DO. Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension 1993;21:618-23. [ Links ]
52. Grassi G, Dell'oro R, Facchini A, Quarti Trevano F, Bolla GB, Mancia G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens 2004;22:2363-9. [ Links ]
53. Masuo K, Mikami H, Ogihara T, Tuck ML. Sympathetic nerve hyperactivity precedes hyperinsulinemia and blood pressure elevation in a young, nonobese Japanese population. Am J Hypertens1997;10:77-83. [ Links ]
54. Egan BM. Neurohumoral, hemodynamic and microvascular changes as mechanisms of insulin resistance in hypertension: a provocative but partial picture. Int J Obes 1991;15 Suppl 2:133-9. [ Links ]
55. Mancia G. Bjorn folkow award lecture. The sympathetic nervous system in hypertension. J Hypertens 1997;15:1553-65. [ Links ]
56. Feihl F, Liaudet L, Levy BI, Waeber B. Hypertension and Microvascular Remodelling. Cardiovasc Res 2008;78:274-85. [ Links ]
57. Adameova A, Abdellatif Y, Dhalla NS. Role Of the Excessive Amounts Of Circulating Catecholamines and Glucocorticoids in Stress-Induced Heart Disease. Can J Physiol Pharmacol 2009;87:493-514. [ Links ]
58. Beilin LJ, Vandongen R, Arkwright Pd, Davidson L. Adrenal and Sympathetic Nervous Activity in Subjects with "Low' and "High' Normal Blood Pressure. J Hypertens 1983;1:13-8. [ Links ]
59. Majewski H, Rand MJ, Tung LH. Activation of prejunctional beta-adrenoceptors in rat atria by adrenaline applied exogenously or released as a co-transmitter. Br J Pharmacol 1981;73:669-79. [ Links ]
60. Borkowski KR, Quinn P. Adrenaline and the development of spontaneous hypertension in rats. J Auton Pharmacol 1985;5:89-100. [ Links ]
61. Borkowski KR. Effect of adrenal demedullation and adrenaline on hypertension development and vascular reactivity in young spontaneously hypertensive rats. J Auton Pharmacol 1991;11:1-14. [ Links ]
62. Brown MJ, Macquin I. Is adrenaline the cause of essential hypertension? Lancet 1981;2:1079-82. [ Links ]
63. Blankestijn PJ, Man in't Veld AJ, Tulen J, et al. Support for adrenaline-hypertension hypothesis: 18 Hour pressor effect after 6 hours adrenaline infusion. Lancet 1988;2:1386-9. [ Links ]
64. Rumantir MS, Jennings GL, Lambert GW, Kaye DM, Seals DR, Esler MD. The' adrenaline hypothesis' of hypertension revisited: Evidence for adrenaline release from the heart of patients with essential Hypertension. J Hypertens 2000;18:717-23. [ Links ]
65. Folkow B. Physiological aspects of primary hypertension. Physiol Rev 1982;62:347-504. [ Links ]
66. Christensen KL, Mulvany MJ. Vasodilatation, not hypotension, improves resistance vessel design during treatment of essential hypertension: a literature survey. J Hypertens 2001;19:1001-6. [ Links ]
67. Mathiassen ON, Buus NH, Larsen ML, Mulvany MJ, Christensen KL. Small artery structure adapts to vasodilatation rather than to blood pressure during antihypertensive treatment. J hypertens 2007;25:1027-34. [ Links ]
68. Buus NH,Bottcher M,Jorgensen CG, et al. Myocardial perfusion during long-term angiotensin-converting enzyme inhibition or beta-blockade in patients with essential hypertension. Hypertension 2004;44:465-70. [ Links ]
69. Kyrou I, Chrousos GP, Tsigos C. Stress, visceral obesity, and metabolic complications. Ann N Y Acad Sci 2006;1083:77-110. [ Links ]
70. Hassellund SS, Flaa A, Sandvik L, Kjeldsen SE, Rostrup M. Long-term stability of cardiovascular and catecholamine responses to stress tests: An 18-Year Follow-Up Study. Hypertension 2010;55:131-6. [ Links ]
71. Davis AM, Natelson BH. Brain-Heart Interactions. The neurocardiology of arrhythmia and sudden cardiac death. Tex Heart Inst J 1993;20:158-69. [ Links ]
72. Gerhardstein BL, Puri TS, Chien AJ, Hosey MM. Identification of the sites phosphorylated by cyclic amp-dependent protein kinase on the beta 2 subunit of l-type voltage-dependent calcium channels. Biochemistry 1999;38:10361-70. [ Links ]
73. Rona G. Catecholamine Cardiotoxicity. J Mol Cell Cardiol 1985;17:291-306. [ Links ]
74. Sordahl LA, Silver BB. Pathological accumulation of calcium by mitochondria: modulation by magnesium. Recent Adv Stud Cardiac Struct Metab 1975;6:85-93. [ Links ]
75. Taam GM, Takeo S, Ziegelhoffer A, Singal PK, Beamish RE, Dhalla NS. Effect of adrenochrome on adenine nucleotides and mitochondrial oxidative phosphorylation in rat heart. Can J Cardiol 1986;2:88-93. [ Links ]
76. Raab W. Adreno-sympathogenic heart disease; Neurohormonal factors in pathogenesis and treatment. Ann Intern Med 1948;28:1010-39. [ Links ]
77. Fleckenstein A, Frey M, Fleckenstein-Grun G. Consequences of uncontrolled calcium entry and its prevention with calcium antagonists. Eur Heart J 1983;4 Suppl H:43-50. [ Links ]
78. Faber JE, Szymeczek CL,Cotecchia S, et al. Alpha1-Adrenoceptor-dependent vascular hypertrophy and remodeling in murine hypoxic pulmonary hypertension. Am J Physiol Heart Circ Physiol 2007;292: H 2316-23. [ Links ]
79. Leicht M, Greipel N, Zimmer H. Comitogenic effect of catecholamines on rat cardiac fibroblasts in culture. Cardiovasc Res 2000;48:274-84. [ Links ]
80.Turner NA,Porter KE,Smith WH, White HL, Ball SG, Balmforth AJ. Chronic beta2-adrenergic receptors timulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc Res 2003;57:784-92. [ Links ]
81. Zhu WZ, Wang SQ, Chakir K, et al. Linkage of beta1-adrenergic stimulation toapoptotic heart celldeath through protein kinase a-independent activation of Ca2+/Calmodulin Kinase II. J Clininvest 2003;111:617-25. [ Links ]
82. Communal C, Colucci WS. The control of cardiomyocyte apoptosis via the beta-adrenergic signaling pathways. Arch Mal Coeur Vaiss 2005;98:236-41. [ Links ]
83. Krizanova O, Myslivecek J, Tillinger A, Jurkovicova D, Kubovcakova L. Adrenergic and calcium modulation of the heart in stress: from molecular biology to function. Stress 2007;10:173-84. [ Links ]
84. Chida Y, Steptoe A. Greater cardiovascular responses to laboratory mental stress are associated with poor subsequent cardiovascular risk status: A meta-Analysis of prospective evidence. Hypertension 2010;55:1026-32. [ Links ]
85. Linden W, Phillips MJ, Leclerc J. Psychological treatment of cardiac patients: a meta-analysis. Eur Heart J 2007;28:2972-84. [ Links ]
86. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology 2005;30:1-10. [ Links ]
Laura Ribeiro
Departamento de Bioquímica Faculdade de Medicina da Universidade do Porto, Alameda Professor Hernâni Monteiro, 4200-319 Porto. Email: lribeiro@med.up.pt

