











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Mastocytosis refers to a heterogeneous group of diseases characterized by atypical mast cell proliferation and accumulation in one or more organs, together with the release of inflammatory mediators with local and systemic effects.1,2
Although the condition’s etiopathogenesis is uncertain, in some cases it is associated with mutations in c-KIT (mast/stem cell growth factor receptor), leading to increased receptor function and consequent proliferation and accumulation of mast cells.1-3 The most commonly described c-KIT mutation is D816V in exon 17, accounting for the substitution of valine for aspartate in codon 816. This mutation is present in 80% of mastocytosis cases in adults and only 35% in children, with the latter having other c-KIT mutations in 40% and no mutation in 25% of cases.4,5 Although c-KIT mutations seem to play a central role in mastocytosis, the fact that some (adult and pediatric) patients present no mutations suggests that other, yet undetermined, factors play a role in clinical presentation.
In childhood-onset mastocytosis (which accounts for approximately two-thirds of cases), the disease usually begins in the first year of life (60-80% of cases), most commonly in the first six months, and the vast majority (80%) refer to cutaneous forms.1,4-6 A systematic review of 1,747 cases reported a preponderance of male gender (male-to-female ratio of 1.4).4
Although a considerable proportion of cases are benign and tend to regress by adolescence, up to 30-50% present with extensive cutaneous forms.1,7 A recently published review reported regression of cutaneous lesions in 67% of cases and stabilization in 27%, with 2-9% of cases having a fatal outcome after a median follow-up of 6 years.4
In adult-onset mastocytosis, other tissues, such as the bone marrow, liver, spleen, lymph nodes, and gastrointestinal tract, can be involved (systemic form), and lesions are monomorphic and smaller in size.1,5 Although rare, systemic forms can also occur in children.4
Case report
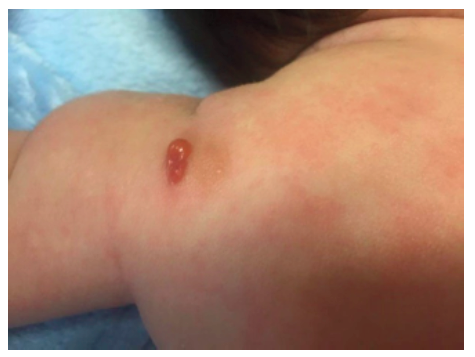
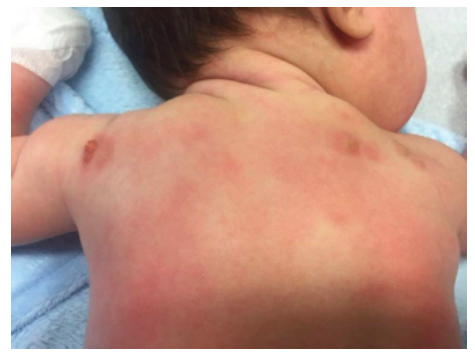
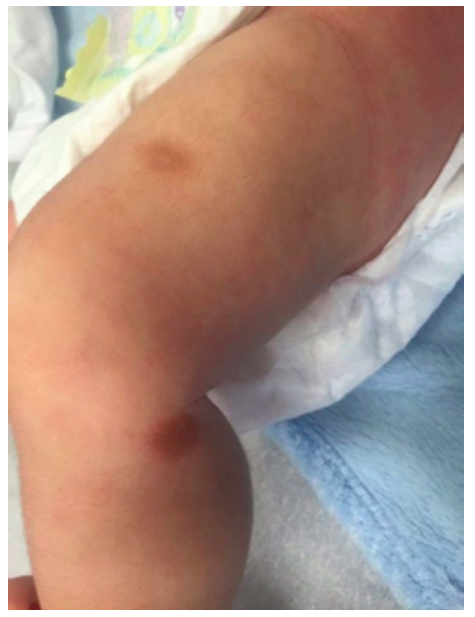
A two-month-old male infant with no relevant medical history was admitted to the Emergency Department with dermatosis with one month of evolution. The condition presented as small-to-medium-sized, tan-to-reddish brown macules (the largest being around 1.5 cm in diameter) and papules, initially distributed over the lower limbs and rapidly spreading to the upper limbs, trunk, face, sparing mucous membranes, scalp, palms, and soles. In the upper and lower limbs, some lesions evolved to nodules and small plaques with around 3 cm of maximum diameter, some with blister formation (Figures 1,2 and 3).
Systemic symptoms, such as dyspnea, vomiting, diarrhea, and acute anaphylactic reactions were excluded. Upon slight lesion scratching or rubbing, a wheal and flare reaction developed, known as positive Darier’s sign. Exposure to physical stimuli, such as pressure (clothes), heat, and sweating, was reported as triggering factors. Lymphadenopathy, hepatosplenomegaly, or fracture signs were not identified.
The initial laboratory study included complete blood count with differential, coagulation profile, C-reactive protein, serum electrolytes, and renal and liver function tests, which revealed no abnormal values. Abdominal and renal ultrasound were normal.
The infant was promptly referred to the Dermatology Department with clinical suspicion of cutaneous mastocytosis.
Despite the characteristic lesions and presence of positive Darier´s sign, a differential diagnosis should be performed in cases of children with macular, nodular, and/or bullous lesions. In the present case, this included bullous impetigo, bullous arthropod bite, linear IgA bullous dermatosis, incontinentia pigmenti, and bullous pemphigoid.7
Skin biopsy was performed in the Dermatology consultation, with histopathological staining with toluidine blue showing a dense inflammatory infiltrate of mast cells in the papillary and reticular dermis. Serum tryptase level was 6 ng/mL (reference value <11 ng/mL). The absence of systemic signs or symptoms, normal serum total tryptase level, and skin biopsy findings (dense infiltrate of mast cells in the dermis forming agglomerations) allowed to establish the diagnosis of maculopapular cutaneous mastocytosis.
The infant was treated with an oral antihistamine agent and topical immunosuppressant (tacrolimus 0.03%) twice daily. Avoidance of triggering factors, such as skin friction of lesions and exposure to hot temperatures, was recommended, including in bath. No recommendations were made regarding other triggering factors not implicated in this case (e.g., foods and medications).
Significant improvement was observed following the implementation of pharmacologic and non-pharmacologic measures, with lesions clearing up and decreasing in number. The patient is currently 25 months (23 months of follow-up) and remains on topical immunosuppressants but has discontinued antihistamines. He presents only tan macules and patches, with no new cutaneous symptoms (blistering lesions, flushes, or itch) since the age of seven months.
Discussion
Based on the 2016 World Health Organization (WHO) revised classification, mastocytosis can be divided into cutaneous, systemic, or localized.8 In the cutaneous form, firstly described in 1869 by Nettleship and Tay, the only affected organ is the skin, and three main presentations can be found: maculopapular cutaneous mastocytosis (or urticaria pigmentosa; 40-70%), solitary mastocytoma (20-50%), and diffuse cutaneous mastocytosis (3-8%).4,8,9
Cutaneous mastocytosis is usually suspected by the presence of typical skin lesions, particularly in children. Darier´s sign is the basic and most important diagnostic test and should be investigated in all patients.5,6 It is more pronounced in children due to the higher concentration of mastocytes in the affected skin.10
Serum tryptase level is a marker of mast cell activity that, when increased (>20 ng/mL), suggests systemic involvement.1,11 It directly correlates with the number of mast cells, including in bone marrow biopsy, and disease severity.1,11,12 Serum tryptase levels tend to decrease with time in concert with improving symptoms. Carter et al. argue that there is value in determining their levels at 6-to-12-month intervals.12
Skin biopsy with histological examination with special stains, such as toluidine blue, giemsa, or monoclonal antibodies to mast cell tryptase or CD117 (KIT), typical shows mast cell infiltrates in a multifocal pattern, confirming the diagnosis.10,13 Eosinophils are commonly found in the dermis, with hyperpigmentation of the basal layer also frequently observed.10
Bone marrow biopsy is usually not recommended in pediatric-onset cutaneous mastocytosis, except in the presence of systemic symptoms, analytical changes (including increased serum tryptase levels), or absence of improvement with prompt treatment.2,6,10 Among patients with high tryptase levels (>20 ng/mL) and severe mediator symptoms, organomegaly appears to be a strong indicator of those who may require bone marrow biopsy.12 WHO proposes a stepwise approach for the diagnosis of the condition, in which patients must have one major criterion (typical skin lesions) and one of two minor criteria (histological ─ multifocal dense infiltrates of mast cells [≥15 mast cells per cluster] or scattered mast cells [≥20 per high microscopic power field]) ─ or molecular ─ point mutation at codon 816 of c-KIT in the affected skin tissue) to be diagnosed with cutaneous mastocytosis.5,10,14
Management of cutaneous mastocytosis includes counseling parents about the disease pathophysiology and the importance of avoiding possible mast cell degranulation triggers.15 Treatment of cutaneous lesions is mainly symptomatic and has only recently been proposed, with no specific treatments available until now.15
Topical medications include glucocorticoids, which may reduce the number of lesions if applied once daily over 8-12 weeks.10 Topical calcineurin inhibitors applied twice daily may reduce mast cell degranulation by inhibiting T-cell activation and cytokine release, offering a safe alternative to glucocorticoids.10
Chronic administration of oral antihistamines (the mainstay of treatment) is often useful in reducing cutaneous and gastrointestinal symptoms, with oral cromolyn sodium specifically used for the latter.10 Other systemic therapies, such as oral corticosteroids or narrow-band ultraviolet B, can also be used in children in selected cases.10
In conclusion, the present report described the case of a two-month-old boy with macular and nodular lesions (some with bullous formation) and positive Darier´s sign, suggestive of cutaneous mastocytosis, which was later confirmed by skin biopsy. The absence of systemic symptoms and normal laboratory tests (including serum tryptase levels) excluded the systemic form of the condition. The use of a topical immunosuppressant, a member of the calcineurin inhibitor class, led to significant lesion improvement a few months later, with no serious adverse effects. Some reports of this treatment can be found in the literature, with the largest case series of 18 patients supporting its use.15 Treatment duration until complete resolution was two to nine months in previously reported cases, and eight months in the above-mentioned largest case series.15 In the present case, a favorable outcome was achieved after 23 months of follow-up. Although the child presented no new lesions after the first five months of treatment, topical treatment was kept due to the association of longer treatment with an increased likelihood of lesion resolution.15
Some predictors of prognosis have been put forward but not fully confirmed, including diagnosis before the age of two years, clinical improvement with treatment, and absence of systemic signs (as in the present case).4,7

