











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Bladder tumors occur more frequently between the fifth and seventh decades of life - they are rare before 40 years of age. Rhabdomyosarcoma is a rare embryonic tumor originating from skeletal muscle tissue.1) When located in the genitourinary tract, it can present with hematuria and anuria.1) Being a tumor of embryonic origin, it is more common in children and very rare in adults: 85% occur in children and adolescents.1
Although most musculoskeletal tumors occur in the chest, arms, and legs, rhabdomyosarcomas often appear elsewhere in the body. The most common site of involvement is the head and neck (30% to 40%), followed by the urinary and reproductive systems (20% to 25%). However, the tumor can appear anywhere in the body, even where there is no skeletal muscle.1 When located in the genitourinary tract, it can present with hematuria and anuria.1
In most cases, bladder tumors are asymptomatic. The clinical condition, when apparent, progresses with hematuria, intermittent or persistent without apparent causes, with an approximate average of 25% of patients presenting macroscopic and 9% microscopic hematuria.1,2 Other manifestations include flank pain, irritative and obstructive symptoms, such as increased voiding frequency, voiding urgency, retention, dysuria, nocturia, hydronephrosis, and changes in renal function. Signs and symptoms vary according to the location and extent of the tumor mass.1,2
Diagnosis consists of vesical visualization through cystoscopy and abdominal and pelvic evaluation through imaging exams, followed by a biopsy or resection.2 It is a very aggressive tumor, which usually requires surgical treatment, with partial resections and even complete removal of a large part of the lower genitourinary tract.2
The presentation of the tumor on ultrasound demonstrates a heterogeneous lesion with sonolucent areas, corresponding to sites of hemorrhage or necrosis. On CT, we should look for an image consistent with a heterogeneous, infiltrative attenuation mass, usually with imprecise borders. On the other hand, MRI reveals a low signal on T1-weighted images and a high signal on T2-weighted images, allowing for a better assessment than ultrasound and CT of the local extension of the tumor and the involvement of adjacent structures.
The nevus sebaceous of Jadassohn (NSJ) is a congenital hamartoma of cutaneous structures, with the epithelial and adnexal origin, present in approximately 0.3% of newborns, and may contain any component of the skin, including sebaceous and apocrine glands or hair.1 Most often seen in the head and neck region, it presents as a well-demarcated yellowish alopecia plaque.1 In our case, the nevus was confirmed with a biopsy and pathological examination of the cephalic lesion. The nature of secondary tumors that arise in sebaceous nevus and the risk of malignant neoplasm are controversial issues.1
When a Jadassohn sebaceous nevus is associated with syndromic features such as mental retardation, central nervous system abnormalities, cardiovascular defects such as the cardiac arrhythmia reported in this case, ocular or skeletal abnormalities, it is called linear sebaceous nevus syndrome, or Schimmelpenning syndrome.1 The etiology is still unknown, but recent studies point to a possible link with the mother´s positivity for the human papillomavirus or mutations in the patched gene (PTCH).2) Luo et al. noticed a novel mutation in the HRAS gene (c.38G>T; p.Gly13Val) was noticed in a mosaic state in NS and rhabdomyosarcoma, which could be a connection between these entities.3
We report a male infant aged four months and 13 days old, weighting 4.3 kg, with a history of diarrhea for ten days, low formula acceptance, and prostration. He had not urinated for four days and had started incoercible vomiting. He was born at 34 weeks, remaining in the neonatal intensive care unit for 48 days due to a previous diagnosis of fetal cardiac arrhythmia.
On physical examination, the baby was hypoactive/reactive, pale 2+/4+ and dehydrated 1+/4+. The abdomen was globular with reduced air-fluid sounds, distended, painful on diffuse palpation, with a thickened spermatic cord, there was a bilateral hydrocele.
He also had large, hairless, yellowish-brown plaques on the scalp (Figure 1), which were already present at birth according to his mother. Histopathological examination of these lesions on the scalp revealed hyperkeratosis, acanthosis, and hyperplasia of the sebaceous glands, compatible with nevus sebaceous of Jadassohn. Karyotype evaluation showed: karyotype 46, XY, presence of variant15CENH+ (increase in centromeric heterochromatin in one of chromosomes 15). Renal function deteriorated - urea (323 mg/ml) and creatinine (7.20 mg/ml).
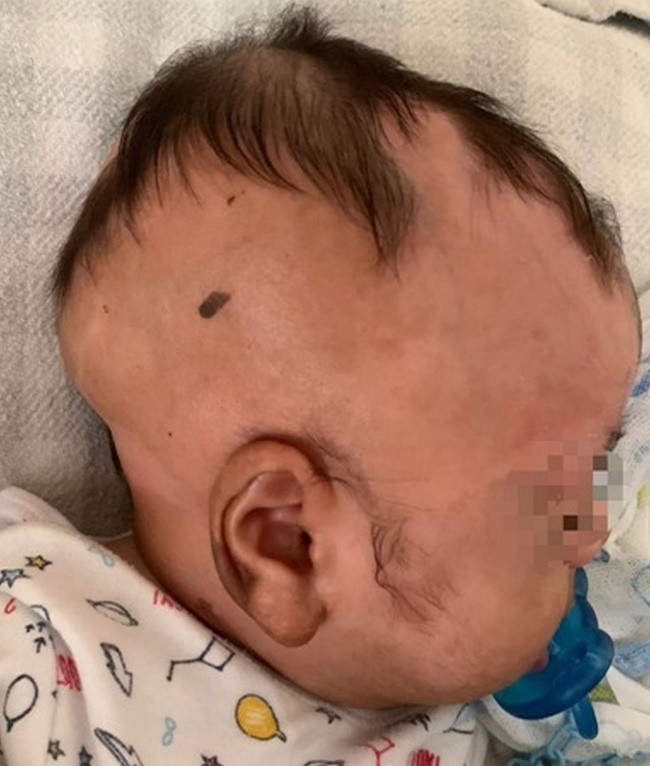
Figure 1: Hairless, yellowish-brown plaques on the scalp of a four-month-old infant. Histopathological examination revealed hyperkeratosis, acanthosis, and hyperplasia of the sebaceous glands, compatible with Jadassohn sebaceous nevus.
Urethrocystography was performed (Figure 2), which demonstrated a defect in the bladder contrast. Ultrasonography and computed tomography (CT) showed a bladder mass measuring approximately 6.5 cm (Figure 2), later confirmed by magnetic resonance imaging (MRI) (Figure 3).
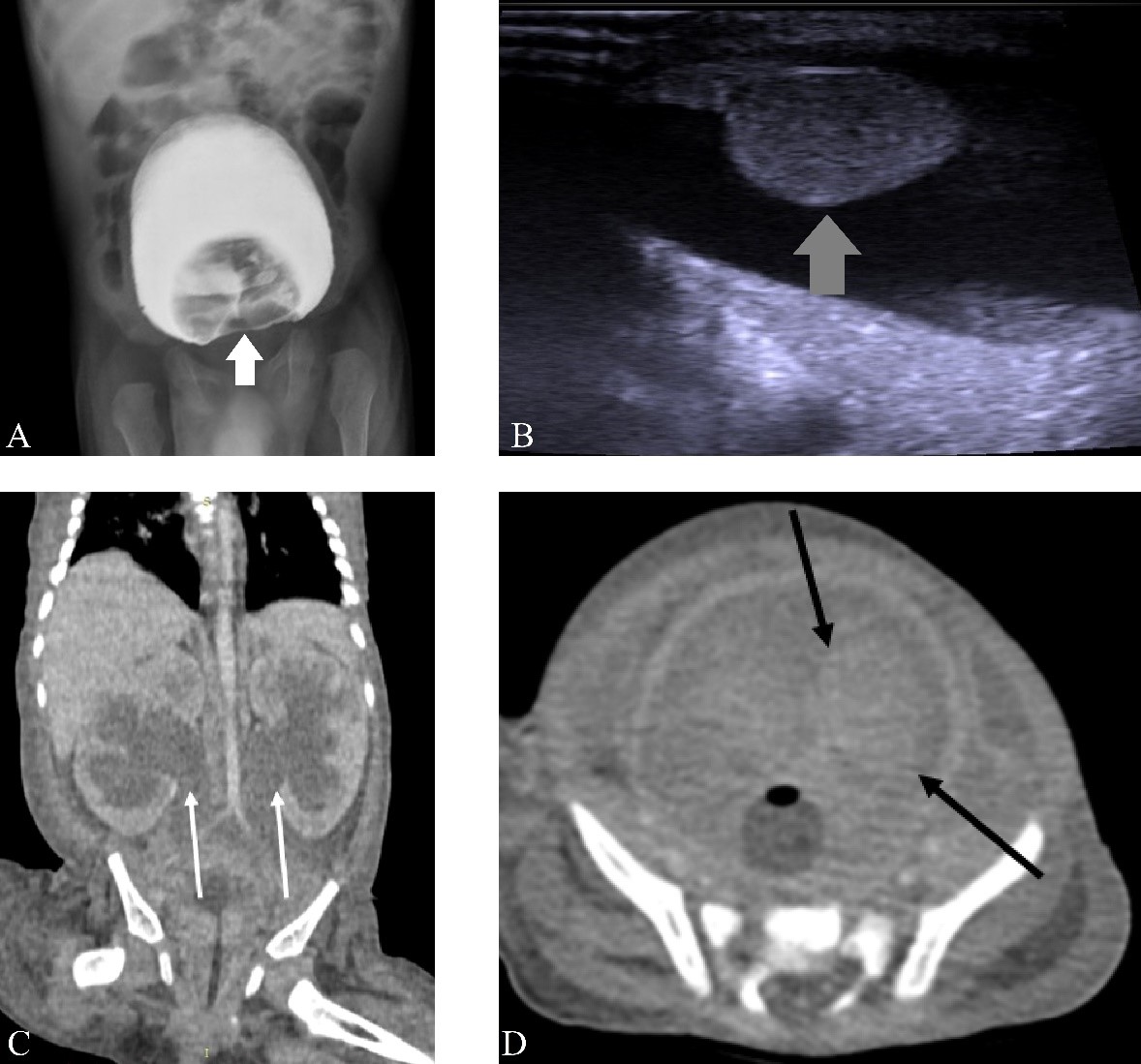
Figure 2: A: urethrocystography showing a defect in the bladder contrast (white arrow). B: Ultrasonography showing expansive and infiltrative lesion affecting the bladder wall (grey arrow). C: contrast-enhanced CT in the coronal section demonstrating bilateral ureterohydronephrosis (white arrows). D: contrast-enhanced CT in the axial section showing an expansive and infiltrative lesion affecting the lateral wall and the vesical floor, invading the ureteral meatus (black arrows), causing bilateral ureterohydronephrosis.
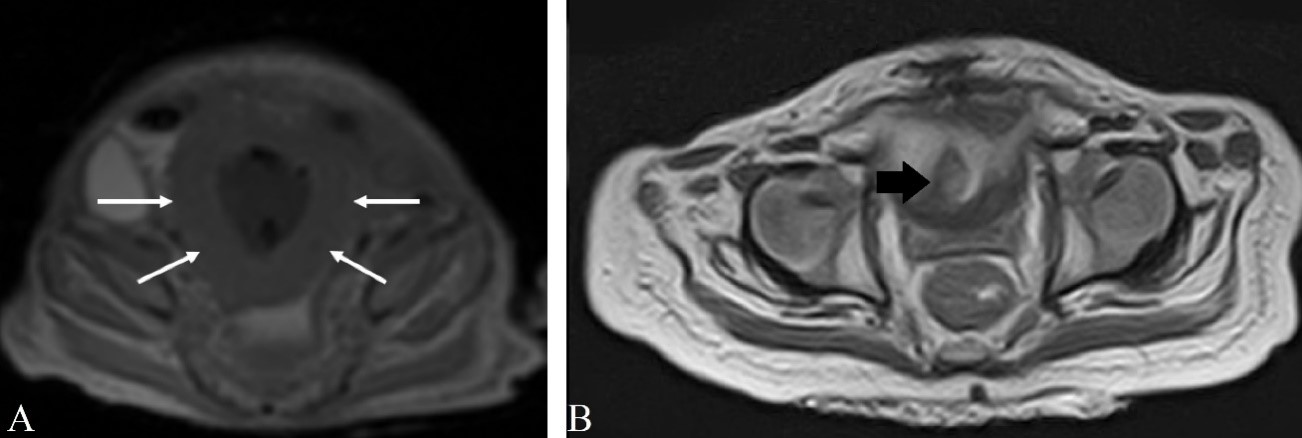
Figure 3: A: MRI in the axial section of the sequence T1 showing diffuse thickening of the bladder walls (white arrows). B: MRI in the axial section of the sequence T2 showing an expansive lesion affecting the bladder wall (black arrow).
The bladder tumor was excised - the anatomopathological analysis diagnosed botryoid-type embryonal rhabdomyosarcoma with compromised margins - and the patient is currently undergoing chemotherapy. Bladder ultrasonography and CT scan were normal after the surgical procedure.

