











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A púrpura de Henoch-Schönlein (PHS) é a vasculite sistémica mais comum em idade pediátrica com atingimento cutâneo, articular, gastrointestinal e renal. Os critérios de diagnóstico foram definidos pela EULAR/PRES (European League against Rheumatism/ Paediatric Rheumatology European Society), nos quais a púrpura palpável é critério obrigatório, associada a pelo menos uma das seguintes manifestações:
Artrite aguda ou artralgia;
Dor abdominal (geralmente difusa, início súbito);
Envolvimento renal (qualquer forma de hematúria ou proteinúria);
Vasculite leucocitoclástica ou glomerulonefrite proliferativa com depósitos de IgA.1
A PHS ocorre predominantemente entre os 3 e os 15 anos de idade e metade dos casos são precedidos por infeção das vias aéreas superiores. A maioria dos casos é autolimitada, com recorrência reportada em 30%.2 As manifestações dermatológicas são caracterizadas por púrpura palpável de predomínio nos membros inferiores, que podem pontualmente evoluir para vesículas e bolhas resultando em necrose cutânea, a qual não apresenta, contudo, valor prognóstico.3
Caso clínico
Reportamos o caso clínico de um rapaz de 3 anos e 8 meses, sem antecedentes de relevo para além de dermatite atópica, que recorreu ao serviço de urgência (SU) por dermatose eritematosa de predomínio nos membros inferiores e febre com dois dias de evolução. História de lesão impetiginada do hálux do pé direito tratada com ácido fusídico tópico três semanas antes do início do aparecimento das lesões cutâneas.
Inicialmente apresentava máculas e pápulas eritemato-purpúricas nas superfícies extensoras dos membros, que evoluíram, em dias (D) 3 de doença, para lesões purpúricas palpáveis nos membros inferiores, associadas a edema bilateral das articulações tibiotársicas e artralgia, com limitação da marcha, mas sem incapacidade. No SU foi feito o diagnóstico de PHS e teve alta com tratamento sintomático (analgésicos), controlo seriado no médico assistente e orientado para consulta de Pediatria. Nas 48 horas seguintes verificou-se aumento progressivo do número de lesões e agravamento do edema articular, sem alteração da tensão arterial (TA) ou da tira-teste de urina (TTU), pelo que manteve apenas vigilância clínica.
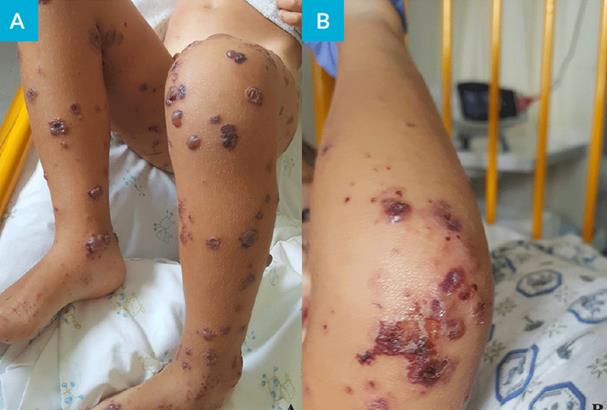
Figura 1: Lesões purpúricas palpáveis dos membros inferiores (A) e membros superiores (B), D10 de doença.
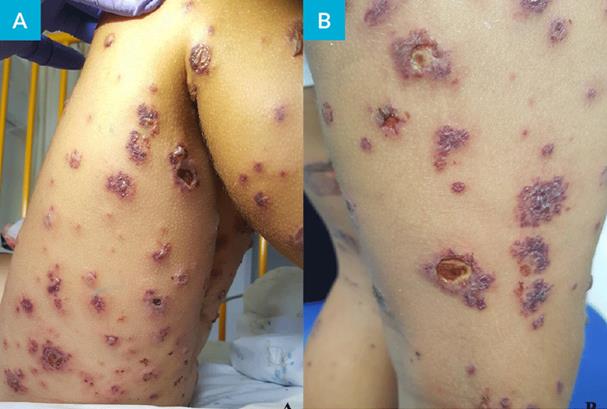
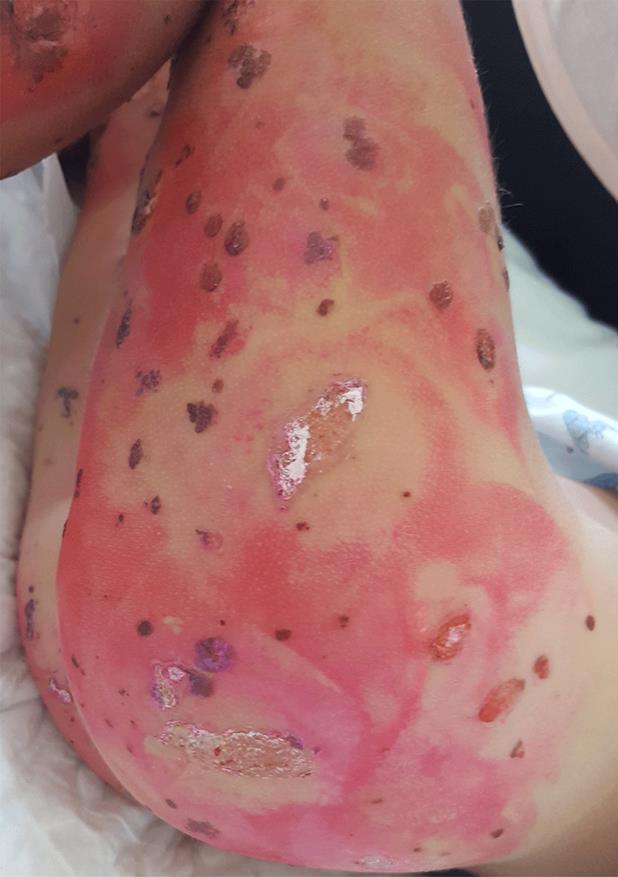
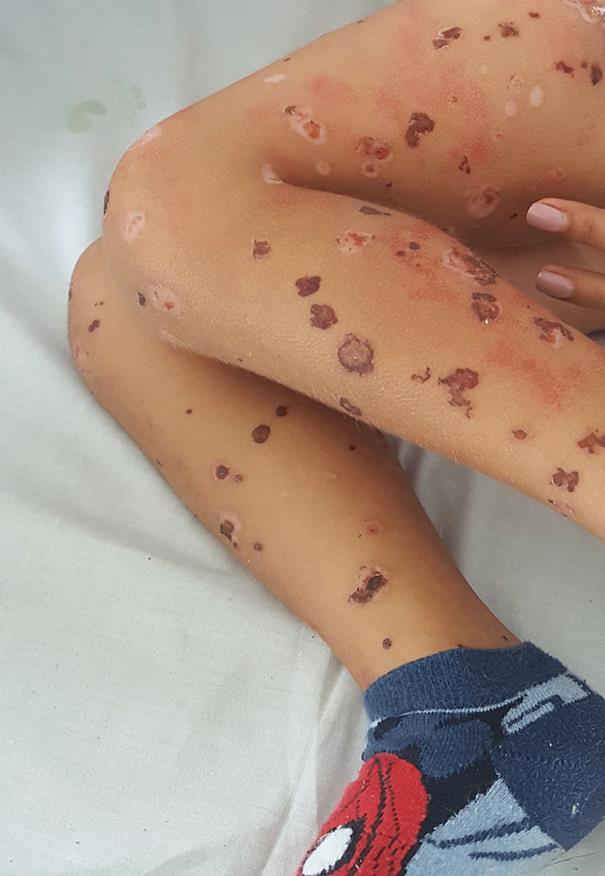
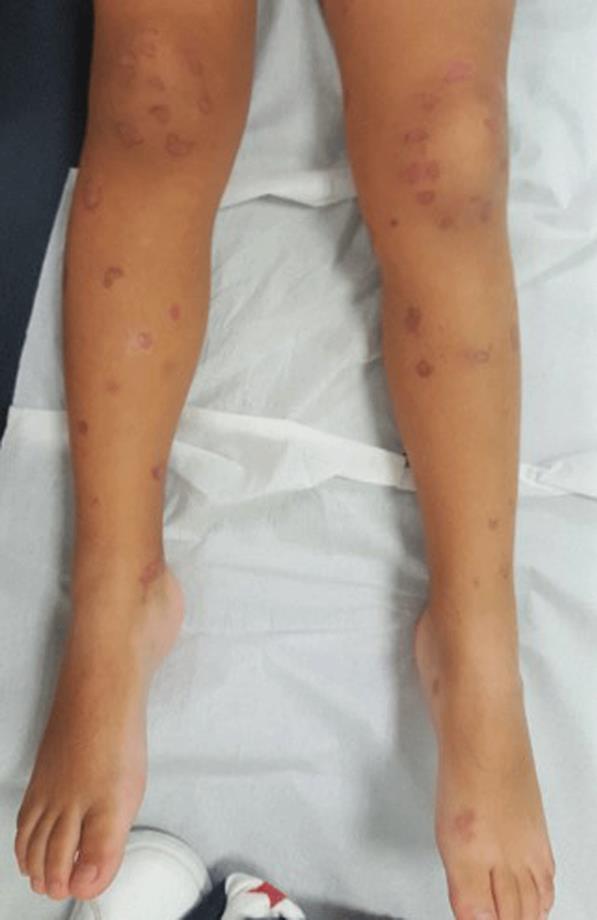
Em D10 de doença mantinha febre com intervalos de apirexia de 4-6 horas, temperatura axilar máxima 39ºC, tendo-se constatado agravamento clínico, com incapacidade total para a marcha e para o ortostatismo, dor abdominal difusa e aumento do número de lesões, com dor exuberante ao toque, o que motivou nova admissão no SU. Ao exame objetivo apresentava TA>P95 para a idade, hiperemia da orofaringe, púrpura palpável com vesículas, bolhas hemorrágicas, necrose cutânea e ulcerações dispersas pelo corpo, com maior predominância nos membros inferiores e glúteos (Fig.s 1 e 2) e edema periorbitário bilateral e dos pés. Sem sinais clínicos de abdómen agudo, hemorragia digestiva ou hematúria macroscópica. O estudo analítico revelou hemoleucograma, função renal, estudo da coagulação e urina tipo II sem alterações, mas PCR 29 mg/L e albumina 3 g/dL. Perante o contexto clínico, e pela suspeita de sobreinfeção cutânea, foi decidido o seu internamento sob amoxicilina/ácido clavulânico endovenoso (EV) 30 mg/kg/dose 8/8 horas, metilprednisolona 1 mg/kg/ dia EV, analgesia com paracetamol e ibuprofeno e ainda morfina antecedendo a realização de cuidados de penso. Manteve-se hemodinamicamente estável, com TA no P50-95 e em apirexia sustentada desde a admissão, com evolução paulatina do quadro cutâneo (Fig.s 3 e 4) e melhoria do edema e do componente articular. Cumpriu 10 dias de antibioterapia e iniciou redução de corticoterapia após cinco dias de tratamento (D15 de doença) que foi suspensa ao 11º dia. Repetiu estudo analítico em D8 de internamento, mantendo hemoleucograma sem alterações, PCR negativa, velocidade de sedimentação 21 mm/h e IgA 213 mg/dL (normal 27-195 mg/dL). Teve alta 48 horas após suspensão do antibiótico, com melhoria das lesões cutâneas, a maioria em fase de cicatrização e sem sinais de sobreinfeção, sem limitação da mobilidade articular dos membros inferiores e sem sinais inflamatórios articulares associados. Foi seguido em consulta de pediatria, mantinha evolução clínica favorável, sem lesões de novo, mantendo áreas cicatriciais hiperpigmentadas (Fig. 5), TA sem alterações e TTU com vestígios de proteínas. Teve alta da consulta dois anos após o episódio inicial, sem outras intercorrências.
Discussão
A PHS é uma vasculite de pequenos vasos, mediada por imunocomplexos IgA. Trata-se de uma vasculite comum em idade pediátrica, com manifestações clínicas variáveis, sendo a púrpura e as artralgias geralmente a apresentação clínica inicial. As erupções bolhosas são manifestações raras, cuja patofisiologia permanece incerta e podem constituir um desafio diagnóstico em doentes com PHS.4 Apesar de considerado um fenómeno raro, a incidência de PHS bolhosa pode estar subestimada pela dificuldade diagnóstica. A presença de vesículas/bolhas não parece ter valor prognóstico, sendo uma apresentação autolimitada, podendo, no entanto, constituir uma comorbilidade importante.
Não existe ainda consenso no tratamento da PHS bolhosa pela falta de estudos e dada a raridade da situação. Alguns estudos apontam para a corticoterapia como forma de reduzir a severidade da dor abdominal e o risco de desenvolvimento de patologia renal, sem interferir, contudo, na probabilidade de recorrência. Alguns autores sugerem que o uso precoce de prednisolona pode reduzir a gravidade e extensão das lesões bolhosas, um pouco pelo seu efeito anti-inflamatório. No entanto, são necessários mais estudos que corroborem a utilização da corticoterapia na PHS bolhosa.2 No caso do nosso doente, a evolução das lesões e as queixas articulares melhoraram após início desta terapêutica. A PHS bolhosa é, regra geral, uma patologia de tratamento em ambulatório. Por vezes, o internamento justifica-se, quer pelo risco de sobreinfeção das lesões, quer pelas comorbilidades associadas, quer pela incapacidade funcional.
A PHS é uma doença autolimitada na maioria dos casos, com recorrência estimada em cerca de 30% dos doentes. O prognóstico depende da severidade do atingimento renal. A maior parte dos estudos refere resolução das lesões cutâneas, com persistência de cicatriz e pigmentação em cerca de 25% dos casos,5 situação presente no doente que manteve lesões cicatriciais e áreas hiperpigmentadas objetivadas nas reavaliações em ambulatório.
Conclusão
A PHS bolhosa é uma entidade rara em idade pediátrica e o seu diagnóstico representa um desafio. A utilização de corticoides pode ser benéfica para alguns doentes, necessitando-se ainda de mais estudos que corroborem a sua utilização. Na generalidade dos casos, a PHS tem um bom prognóstico.

