











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Images in Case
We present a male patient, in his twenties, with established diagnosis of Mucopolysaccharidosis type VI by molecular genetic analysis with detection of a homozygotic mutation in arylsulfatase B (ARSB) gene, elevated levels of urinary glycosaminoglycan and demonstrated reduced arylsulfatase B (ASB) enzyme activity in isolated leukocytes and fibroblasts. He has been on enzyme replacement therapy with galsulfase ever since he was diagnosed at the age of 7. As main clinical features he has short stature and hypogonadism, treated with testosterone replacement therapy. He also suffers from moderate mitral and aortic valve insufficiency, controlled with a combination of furosemide, spironolactone and lisinopril. No other symptoms are reported. Regarding his surgical history, he was submitted to bilateral pelvic and femoral osteotomy with 18 months due to hip dysplasia, bilateral carpal tunnel surgical release to treat bilateral carpal tunnel syndrome, as well as bilateral corneal transplantation surgery as he had severe corneal clouding.
A skeletal radiographic evaluation was requested to monitor disease progression, in which typical radiological features were found (displayed on Figures 1, 2 and 3).
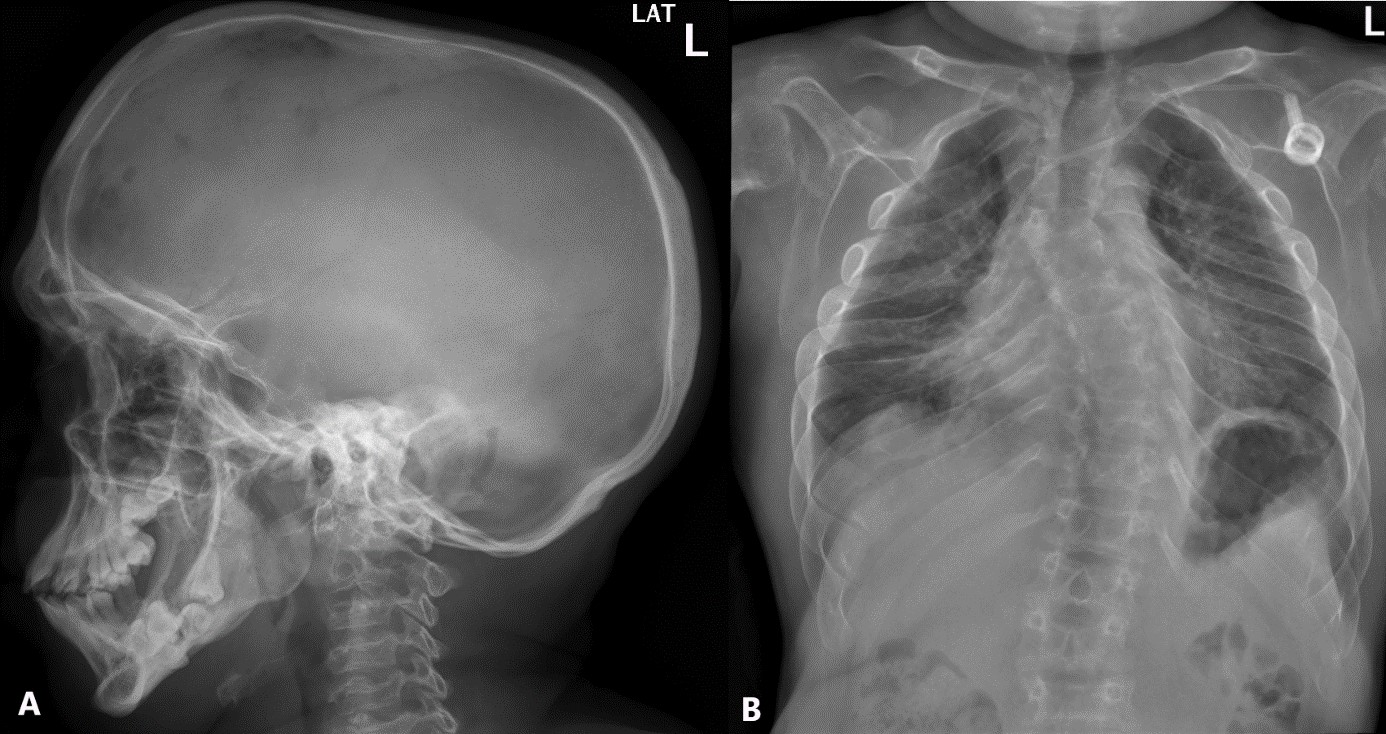
Figure 1: Lateral skull radiograph (A) with macrocephaly, with widening of both transversal and antero-posterior diameters, “J-shaped” sella turcica, discrete thickening of cortical bone, prognathism and widely spaced teeth; Frontal chest radiograph (B) showing short and thickened clavicles, “paddle-shaped” ribs with anterior enlargement and posterior funnelling.
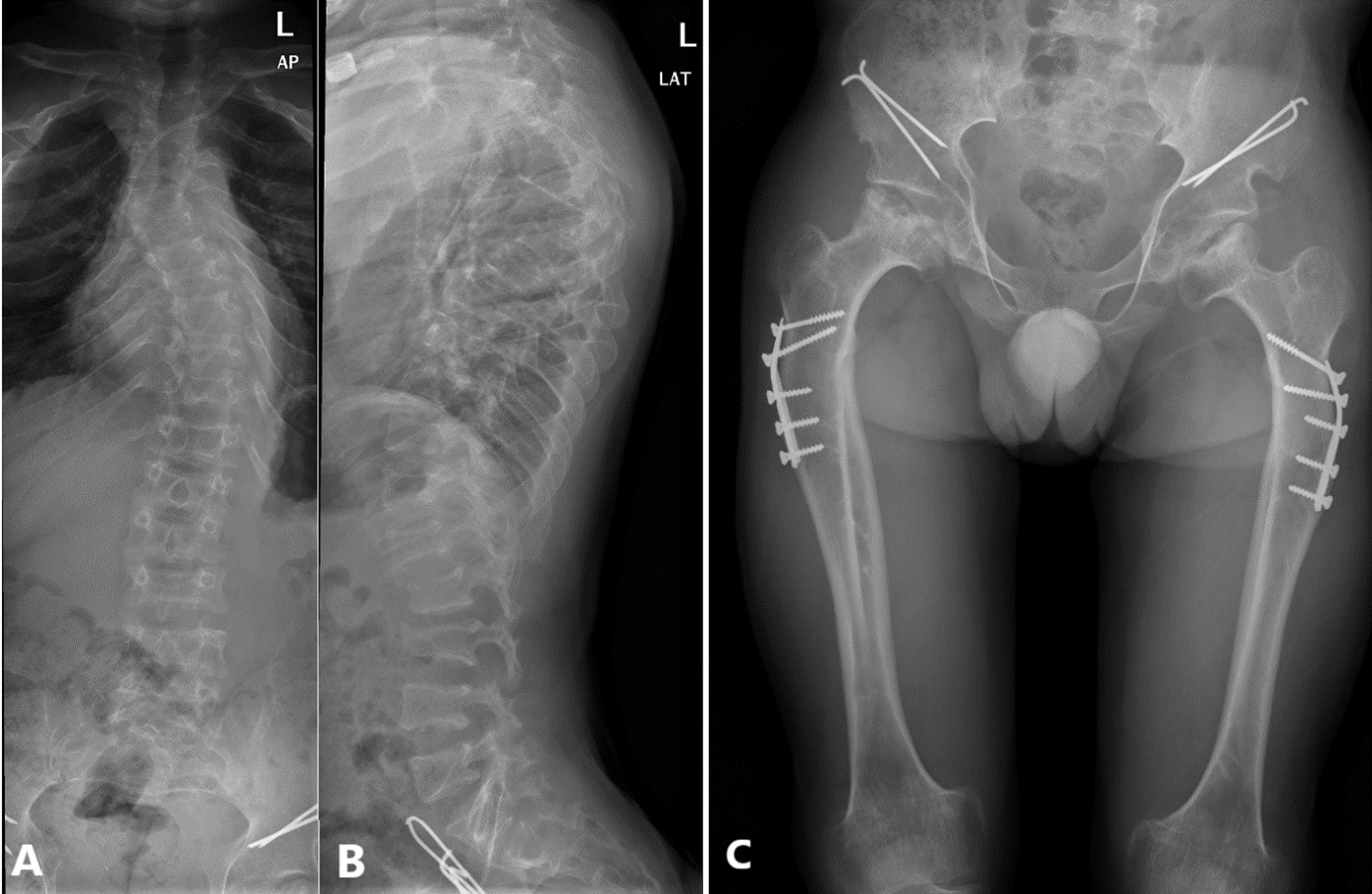
Figure 2: Anteroposterior (A) and lateral (B) radiograph of the dorso-lumbar spine exhibiting “wedge-shaped” vertebral bodies, elongated vertebral pedicles, severe dextro-convex dorso-lumbar scoliosis producing dorsal gibbus deformity, straightening of the physiological lumbar lordosis; Anteroposterior hip and femoral radiograph (C) displaying rounded hypoplastic iliac wings, flat acetabular roof, with signs of surgical material related to hip dysplasia corrective surgery as well as long and narrow femoral neck, dysplastic femoral head, broad proximal femoral diaphysis.
Discussion
Mucopolysaccharidosis type VI, also known as Maroteaux-Lamy syndrome, is a multisystem disease, caused by a mutation in the ARSB gene, leading to an autosomal recessive lysosomal storage disorder due to deficiency of ASB and associated accumulation of glycosaminoglycans, specifically dermatan-sulphate. Incidence rate per total live births in Portugal is estimated in 1 in 238,095, which illustrates the slight relative increase in Mucopolysaccharidosis type VI frequency comparing to other types of mucopolysaccharidosis in Portuguese (16%) and Brazilian (18,5%) population. This can be partially explained by an uprisen prevalence of specific mutations, such as 1533del23, present in 23% of alleles amidst Brazilian Mucopolysaccharidosis type VI patients, which is also found in Portuguese patients, with unknown frequency. However, no specific ethnic group has been associated with a heightened risk of this syndrome.1
Maroteaux-Lamy syndrome can have numerous different clinical findings, with highly variable genetic findings, as well as phenotypic expression.1
Being a multisystem disease it has many clinical manifestations such as organomegaly, with hypertrophic cardiomyopathy and hepatosplenomegaly, cardiac valve dysfunction, respiratory symptoms, as these patients may suffer from reduced pulmonary function and sleep apnoea, ophthalmologic impairment, with corneal clouding and photophobia, as well as hearing disorders, with hearing loss due to repeated otitis media, sinusitis and also other symptoms such as inguinal or umbilical hernia and carpal tunnel syndrome.1,2
Maroteaux-Lamy syndrome exhibits characteristic skeletal chondrodystrophic abnormalities generally referred to as dysostosis multiplex, which are not exclusive to this disease, and can be found in other types of mucopolysaccharidosis, mucolipidoses and other storage diseases.1,2 In this case our patient showed classic radiological features of Mucopolysaccharidosis type VI on skeletal radiographic evaluation.

