











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Reed's syndrome is a rare genodermatosis, characterized by the presence of multiple cutaneous and uterine leiomyomatosis (MCUL) in women. This syndrome can be associated with renal cell carcinoma, being labeled as hereditary leiomyomatosis and renal cell carcinoma (HLRCC), and leiomyosarcoma. Both MCUL and HLRCC have an autosomal dominant pattern of inheritance. They result from a germline heterozygous mutation of fumarate hydratase (FH) gene on chromosome 1q42.3-q43. It is thought that just a few specific mutations are associated with the development of renal cell cancer. FH gene encodes the fumarate enzyme, which converts fumarate to malate in the Krebs cycle and is classified as a tumor suppressor gene.1
Case Report
A 55-year-old woman presented to routine consultation with her family doctor. The patient had no current diseases and was not taking any medication regularly. The physical examination was normal, except for the presence of multiple papules and nodules on her chest (Fig. 1) and left arm (Fig. 2). They were asymmetrical, irregular, skin-colored, with a smooth surface, and she reported that they were sometimes associated with pruritus and had been present for about two decades, slowly increasing over the years.

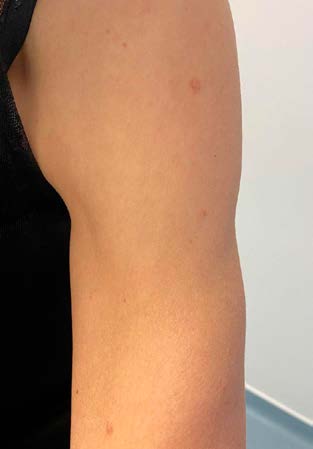
The patient's past medical history was significant for myomectomy thirty years ago, and hysterectomy eight years ago, due to uterine leiomyomas. She was unaware of the family history. A papule biopsy from her chest was taken, and the histological examination revealed a leiomyoma. Clinical and histological findings, combined with surgical history, suggested Reed's syndrome. Therefore, a genetic test was conducted. The result showed a heterozygous variation of FH gene, confirming the Reed's syndrome diagnosis.
Given this, the patient underwent tomographic examination, which showed a renal cyst with a 13 mm diameter. No evidence of renal carcinoma was found. Her parents and siblings were also referred for genetic counseling.
Discussion
Reed’s syndrome is rare, but it has characteristic clinical expressions that all doctors must be aware of because of its’ multisystem involvement. When suspected, genetic testing and counseling should be offered to the patients, and when confirmed, also to their family members.
When a patient presents with cutaneous leiomyomas, it is important to perform a complete history, including family and surgical antecedents, and physical examination. Although not compatible with our patient’s history, cutaneous leiomyomas are usually the first manifestation of the disease, with a mean age of 25 years old.2 Cutaneous leiomyomas or piloleiomyomas appear as small (0.5-2 cm) firm skin colored papules or nodules. They arise from the arrector pili muscles, which are responsible for making hairs stand on end. The papules and nodules can be asymptomatic or associated with pain and paresthesias.3 They are most frequently located in extensor surfaces of the extremities, but also on the trunk, neck and face. These lesions develop often in clusters, commonly in a segmental distribution, and tend to increase in number and dimension with aging.4
On the other hand, the diagnosis’ mean age of uterine leiomyomas is 30 years old.2 Women can experience several gynecological symptoms such as dysmenorrhea, menstrual irregularities, menorraghias and fertility problems, which emphasize the importance of an early diagnosis, allowing a proper counseling to female patients.5
Although it is rare, leiomyomas can degenerate into leiomyosarcomas.6) Treatment of cutaneous leiomyomas depends on the number, extension, and symptomatology. Camouflage with cosmetics and avoidance of cold may be sufficient for some patients.7 For single and small lesions, surgical excision, cryotherapy, carbon dioxide laser ablation and electrocoagulation are an option.4,8 For more symptomatic leiomyomas, calcium channel blockers (particularly nifedipine), an alpha-adrenoceptor blocker (phenoxybenzamine) and gabapentin have been used with limited effectiveness.7,9-11
Renal cell carcinoma is the most concerning feature of the heterozygous mutation of FH gene. This renal cancer does not occur in all individuals with the disease, but unfortunately, when present, it occurs at an early age (median of 44-year-old) and it is usually aggressive. Papillary type 2 renal cell carcinoma is the most frequent subtype. The patient can be asymptomatic or can complain about lumbar pain and hematuria. Annual renal imaging should be recommended to anticipate the detection and treatment of renal cell carcinoma, and magnetic resonance imaging and/or computerized tomography are the preferred exams.12 The incidence of benign renal cysts seems to be higher in HLRCC patients (36%), compared to the general population (4.6%-8.2%).13
This case emphasizes the importance of systematic review of skin lesions, as they can be the diagnostic clue for systemic diseases. An early diagnosis enables the genetic counseling of both the patient and his family members, through a multidisciplinary approach that allows prompt fertility counseling to female patients and early detection of the underlying malignancies - renal cell carcinoma and leiomyosarcoma.
The article was submitted for presentation at WONCA Europe Conference 2022 and is awaiting approval.

