











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introducción
La atresia aural congénita (AAC) es un defecto congénito que se presenta en 1 de cada 10000 a 15000 recién nacidos vivos. Puede presentarse de forma aislada o asociada a un síndrome polimalformativo, entre los que se incluye al síndrome de ojo de gato. Presentamos un caso clínico de un paciente con síndrome de ojo de gato en el cual destacan las malformaciones otológicas asociadas que se documentan radiológicamente.
DESCRIPCIÓN DEL CASO
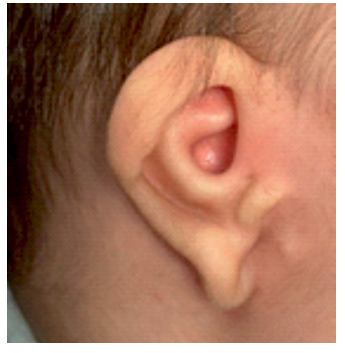
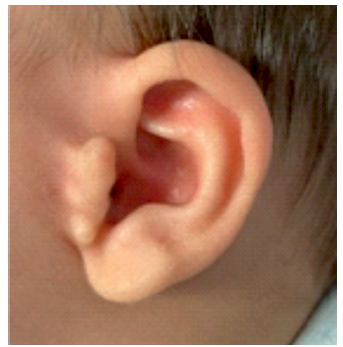
Se presenta el caso de un varón nacido a término con presencia de atresia anal, microtia bilateral grado I con agenesia de canal auditivo externo (CAE) derecho (imagen 1 y 2) y rasgos faciales dismórficos consistentes en fisuras palpebrales con desviación inferior, hipertelorismo y labio superior fino. No hay alteraciones a otros niveles. El embarazo y el parto trascurrieron sin incidencias. Hay constancia de consanguineidad parental de cuarto grado. A los 4 meses de edad el paciente es remitido a nuestro centro para la realización de estudios diagnósticos y manejo terapéutico.
El estudio audiológico mediante potenciales evocados auditivos de vía aérea (VA) y ósea (VO) demostró la existencia de una hipoacusia bilateral de transmisión de grado moderado-severo con umbrales de 60-70 dB en VA y 10 dB en VO (imagen 3). Potenciales evocados troncoencefálicos realizados mediante transductor Sentiero Advanced (SOH100360) con auricular EC-03 (VA) y B71 (VO) ambos con estímulo “chirp” alternante. No se realizaron otoemisiones acústicas debido a las malformaciones craneofaciales y las alteraciones en ambos conductos auditivos externos.
El estudio genético del cariotipo en sangre periférica demostró la presencia de un microcromosoma supernumerario bisatelitado que implica una triplicación parcial en mosaico de la región proximal en el brazo largo del cromosoma 22 (22q11). El cariotipo que presenta es: 47,XY,+mar[16]/46,XY[9] (imagen 4), alteración genética responsable del síndrome de ojo de gato (SOG).
La tomografía computarizada (TC) de peñascos mostró una agenesia de CAE derecho en sus porciones membranosa y ósea y una estenosis severa de la porción membranosa del CAE izquierdo con ocupación por tejido con consistencia de partes blandas en el fondo del mismo (imagen 5). Además, se describe una displasia de cadena osicular bilateral con fusión de martillo y yunque de manera bilateral (imagen 6), asociando también en oído derecho una fusión de ambos a la pared lateral de la cavidad timpánica. Se localizan ambos estribos (imagen 7). Se observó una hipoplasia de celdillas mastoideas y de cavidad timpánica bilateral (imagen 6). Se apreció también una dilatación del conducto subarcuato bilateral (imagen 8). No hay presencia de alteraciones a otros niveles. Tanto cócleas, vestíbulo y canales semicirculares están indemnes. Recorrido normal de ambos nervios faciales (imagen 9)
Se realizó una adaptación bilateral de audífonos de vía ósea acoplados con una banda elástica y apoyo logopédico que obtuvo buena respuesta auditiva. En el futuro, cuando la edad y las condiciones del paciente lo permitan, se planteará la adaptación de implantes auditivos de vía ósea.
El manejo diagnóstico y terapéutico integral del paciente fue consensuado en un comité multidisciplinar con presencia de pediatras, cirujanos pediátricos, otorrinolaringólogos y cirujanos maxilofaciales.
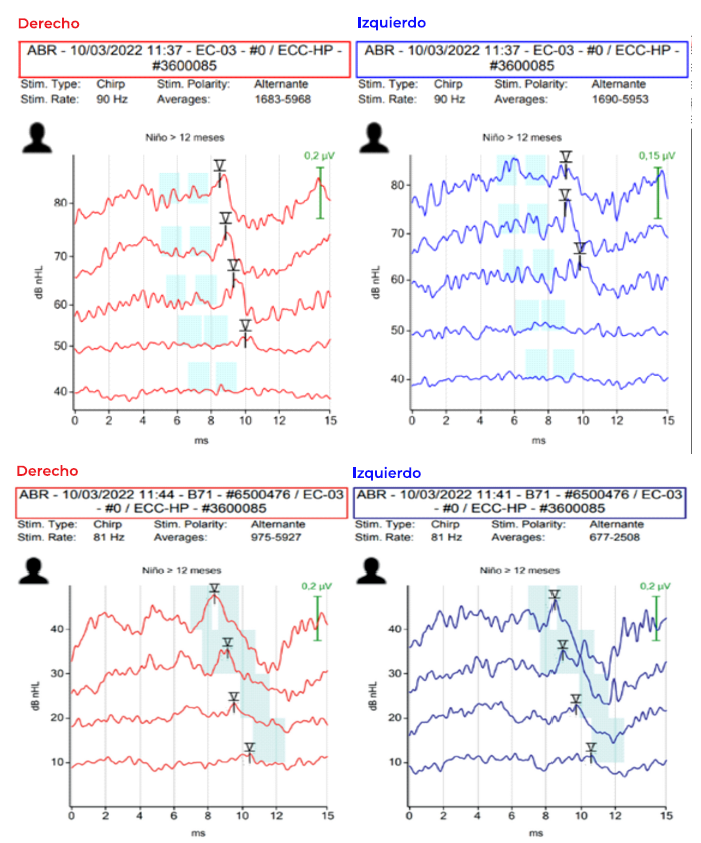
Imagen 3 Potenciales evocados de troncoencéfalo. A: vía aérea con umbrales de 60 dB en oído derecho y 70 dB en oído izquierdo y aumento de latencia en todas las ondas en ambos oídos. Transductor EC-03, estímulo “chirp” alternante. B: vía ósea con umbrales de 10dB. Transductor B71, estímulo “chirp” alternante
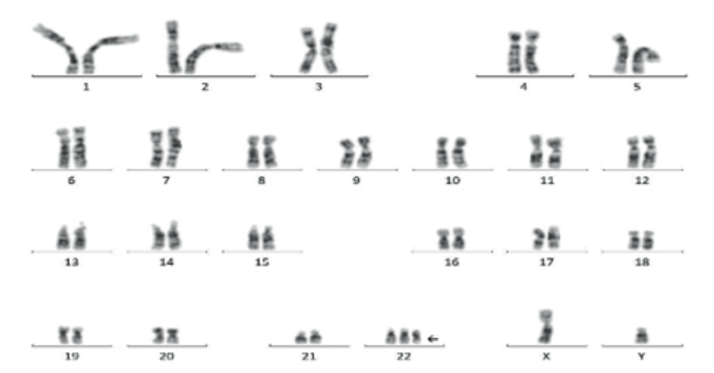
Imagen 4 Cariotipo en sangre periférica 47,XY,+mar[16]/46,XY[9]. Se señala microcromosoma supernumerario bisatelitado (flecha)
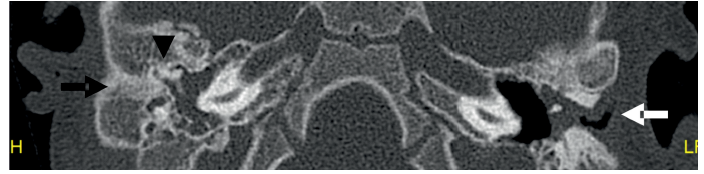
Imagen 5 Imagen transversal de TC identificando atresia CAE derecha (flecha negra) y estenosis de porción membranosa de OI (flecha blanca), se aprecia aireación parcial de caja timpánica izquierda y ocupación completa de la derecha. Se aprecia fusión martillo y yunque a pared anterior cavidad timpánica (cabeza
de flecha).
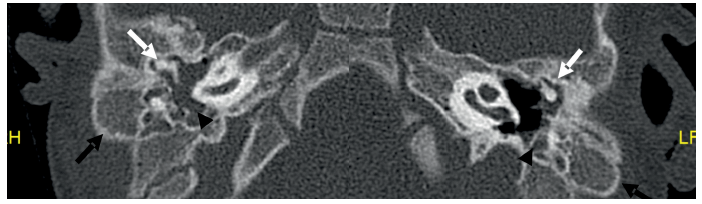
Imagen 6 Se observa displasia de cadena bilateral con fusión martillo y yunque (flechas blancas). Hipoplasia de celdillas mastoideas (flechas negras). Hipoplasia de cavidad timpánica bilateral con tejido de partes blandas en su interior (cabezas de flecha).
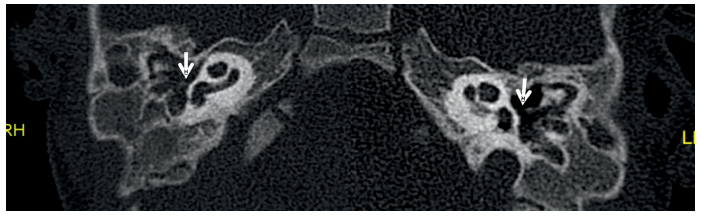

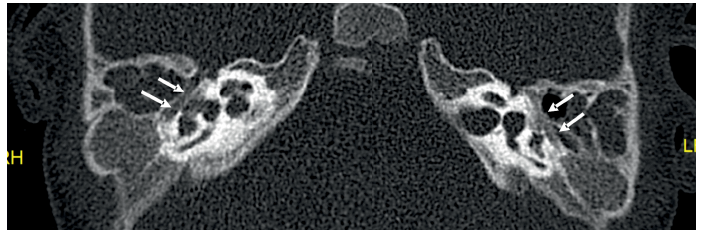
Imagen 8 Imagen transversal de TC en ventana ósea. Se identifica dilatación bilateral y simétrica de los dos tercios laterales de canales subarcuatos o petromastoideos (flechas blancas) con preservación del tercio proximal (flechas negras).
Discusión
El SOG es un síndrome genético raro causado por la trisomía o tetrasomía parcial del cromosoma 22. Tiene una incidencia de 1: 150.000 nacidos vivos 1. Se caracteriza clínicamente por la combinación de coloboma de iris, atresia anal, fisuras palpebrales con inclinación inferior, apéndices preauriculares, malformaciones cardiacas y renales e inteligencia normal o límite2. El SOG presenta una elevada variabilidad fenotípica incluso entre individuos afectos de una misma familia2. Dicha variabilidad condiciona la situación de infradiagnóstico de esta entidad 3,4.
Entre las malformaciones auriculares descritas se encuentran la oreja de implantación baja, el apéndice o fístula preauricular, el lóbulo hipoplásico, el antihélix prominente y la atresia de CAE 1. Como podemos observar la mayor parte de anomalías auriculares que encontramos en la literatura1 son malformaciones menores, en nuestro caso en particular encontramos, además de la atresia de CAE, alteraciones a nivel del oído medio objetivadas ampliamente mediante TC.
En el SOG es habitual la presencia de un cromosoma supernumerario bisatelitado derivado del cromosoma 22 que condiciona la duplicación o triplicación de las secuencias proximales del brazo largo de dicho cromosoma (22q11). También ha sido descrita la duplicación intersticial de la región 22q11. En algunas ocasiones, y así fue en nuestro caso, hay presencia de mosaicismos del cromosoma supernumerario.
En la literatura no hay consenso sobre si existe correlación entre la severidad fenotípica y el grado de mosaicismo. Algunos autores postulan que no existe tal correlación 3, sin embargo, un estudio reciente incide en la existencia de la misma5.
Los casos tienden a ser esporádicos aunque afectaciones de patrón familiar han sido descritas 6. La realización de un cariotipo es la prueba diagnóstica de referencia. También se pueden realizar técnicas de hibridación fluorescente in situ (FISH) o un array genómico de hibridación7.
Es esencial el tratamiento integral del paciente consensuado en comités multidisciplinares para solventar las diferentes malformaciones presentes en el paciente8. Así fue en el caso que nos ocupa, se decidió tratar en primer lugar la malformación anorrectal por ser la más urgente y posteriormente se completó el estudio y tratamiento del resto de malformaciones.
El pronóstico tiende a ser favorable pero depende fundamentalmente de la presencia o no de malformaciones renales y cardiacas asociadas 4. Existen casos prenatales de gran severidad cuyo pronóstico depende de la hipoplasia cerebelosa9.
El SOG es raro pero debe ser sospechado ante la presencia conjunta de malformaciones anorrectales, auriculares y oculares. El estudio genético mediante cariotipo es fundamental para la confirmación diagnóstica mientras que el estudio audiológico es esencial para filiar correctamente la afectación auditiva y poder llevar a cabo estrategias terapéuticas precisas y personalizadas.
Conflicto de intereses
Los autores declaran que no tienen ningún conflicto de interés con respecto a este artículo.
Confidencialidad de los datos
Los autores declaran que siguieron los protocolos de su trabajo en la publicación de datos de pacientes.
Protección de personas y animales.
Los autores declaran que los procedimientos seguidos están de acuerdo con las normas establecidas por los directores de la Comisión de Ética e Investigación Clínica y de acuerdo con la Declaración de Helsinki de la Asociación Médica Mundial.

