











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
In the year 2020, Beck et al. introduced a groundbreaking autoinflammatory syndrome known as VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic)1. This syndrome represents a recent discovery that has forged connections between previously unrelated inflammatory disorders and serves as an exemplar for a new class of hemato-inflammatory diseases2. VEXAS syndrome is an acknowledged monogenic autoinflammatory disorder that predominantly emerges in adulthood, typically in the fifth decade of life or later, and demonstrates a marked male predominance3.
While the precise prevalence of VEXAS syndrome remains uncertain, it is noteworthy that more than 200 cases have been documented in the medical literature, suggesting that it may be more prevalent than originally believed2. This condition is characterized by somatic mutations in the X-chromosomal UBA1 gene, which plays a pivotal role in maintaining protein homeostasis within hematopoietic progenitor cells1. It is marked by profound and relentless systemic inflammation, often presenting as a severe and progressively deteriorating clinical condition that is unresponsive to conventional therapeutic modalities (namely methotrexate, mycophenolate mofetil, and azathioprine)2.
The clinical manifestations of VEXAS syndrome can vary, with systemic inflammation predominantly impacting the skin, lungs, blood vessels, and cartilage. Key clinical features encompass the presence of fever, the presence of vacuoles in neutrophilic and erythroid precursors, elevated inflammatory markers, and cytopenias3. Dysplastic changes in the bone marrow may manifest as macrocytic anemia and thrombocytopenia and could eventually progress to a myelodysplastic syndrome3. Cutaneous symptoms typically present as Sweet-like multiple, firm, tender, infiltrated erythematous or violaceous papules, nodules, and plaques affecting the trunk, limbs, and neck. Notably, cutaneous signs are part of the initial presentation in approximately 63% of patients. Dermatologists frequently assume a pivotal role in the diagnostic process of VEXAS syndrome, given that cutaneous manifestations can serve as indicators of potential systemic complications2. The relentless inflammation observed in VEXAS syndrome leads to symptoms such as fever, fatigue, weight loss, lymphadenopathy, pulmonary infiltrates with neutrophilic alveolitis, pulmonary vasculitis, venous thromboembolism, inflammatory arthritis, synovitis, serositis, as well as ophthalmic complications such as uveitis and scleritis. Disease-related mortality varies between 40 and 63% in different case series, consistently reflecting high mortality rates with substantial associated morbidity1.
The objective of this article is to present a case of VEXAS syndrome with the aim of increasing awareness about this recently identified medical condition.
Clinical case
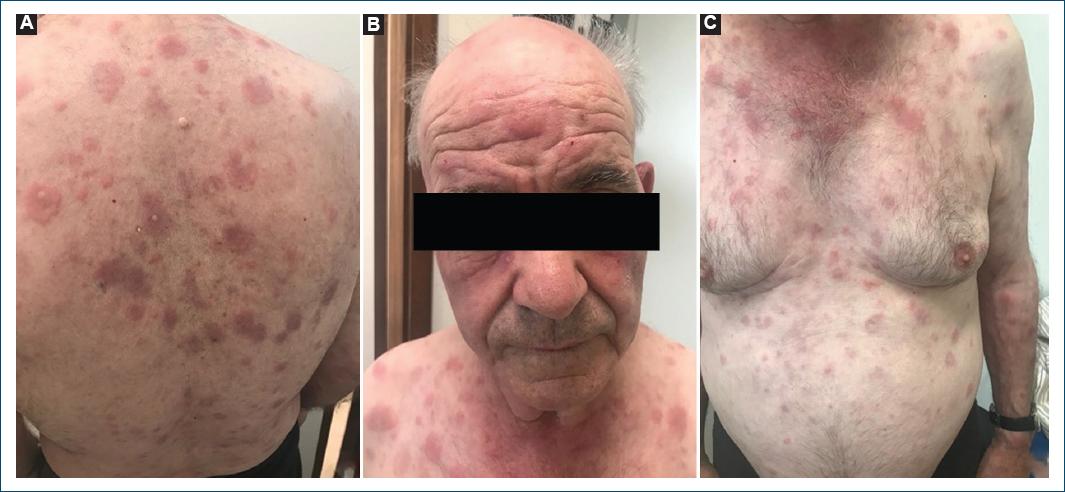
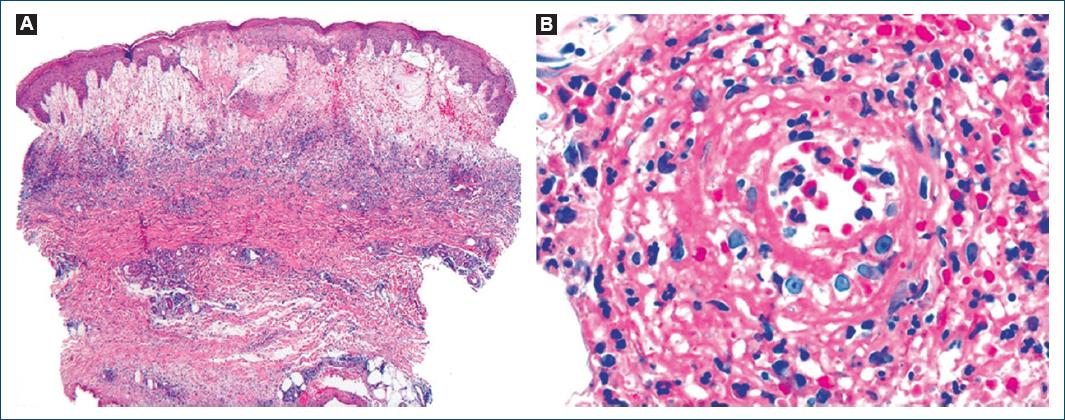
We describe the case of a 78-year-old man, with a personal medical history of type 2 diabetes mellitus, hypertension, and dyslipidemia. He had no significant family medical history and reported no allergies to medications. At the age of 72, he presented with a 2-month history of pruritic, erythematous-violaceous, tumescent, annular plaques on his face, back, and limbs (Fig. 1). There was no recent history of medication use, and systemic symptoms were absent. The initial diagnostic considerations included Sweet syndrome, urticarial vasculitis, cutaneous lymphoma, and lupus tumidus. Consequently, a skin biopsy was performed for histopathological examination (HE staining). The skin biopsy revealed a dermal papilla edema and a perivascular and interstitial dermal inflammatory infiltrate primarily composed of neutrophils, characterized by intense karyorrhexis (leukocytoclasia) with the participation of occasional lymphocytes and xanthomatous macrophages (Fig. 2). Further investigations were conducted, including a complete blood count, peripheral blood smear, age-appropriate oncological screening, serological evaluations, Interferon Gamma Release Assay, blood and urine protein electrophoresis, antinuclear antibodies (ANA) testing, anti-SS-A and anti-SS-B antibodies, rheumatoid factor assessment, complement studies, and peripheral blood cytometry. These investigations did not reveal clinically significant abnormalities. The patient was treated with prednisolone 1 g/kg/day, followed by hydroxychloroquine 400 mg/day, dapsone 100 mg/day, and salazopyrin up to 2 g/day, sequentially with primary and/or secondary failure. Given the recurrent and refractory nature of the dermatosis, repeat skin biopsy for histopathological examination and flow cytometry were performed, yielding results consistent with the initial biopsy. At 74 years of age, the patient concurrently developed bilateral panuveitis, which demonstrated improvement with corticosteroid therapy. Subsequently, a progressive constitutional syndrome emerged, associated with monoclonal gammopathy of uncertain significance (characterized by an IgM/kappa monoclonal protein peak) and macrocytic anemia. Myelogram revealed evidence of myelodysplasia. Thoracic, abdominal, and pelvic computed tomography revealed no pertinent clinical alterations. Considering these clinical and laboratory findings, the patient’s advanced age, and his male gender, the possibility of VEXAS syndrome was entertained. Peripheral blood genetic testing involving direct sequencing of exon 3 of the UBA1 gene conclusively verified somatic mosaicism within the gene, elucidating the presence of the Met41Leu (c.121A > C) variant and, consequently, confirming the diagnosis. Therapeutic intervention included systemic corticosteroid therapy and methotrexate, which elicited a partial response. However, the patient succumbed to sepsis with a respiratory onset.
Discussion
The identification of a potential shared genetic basis for inflammatory skin conditions offers valuable insights into the underlying mechanisms governing cutaneous inflammation2.
In nearly all individuals diagnosed with VEXAS syndrome, specific missense mutations have been pinpointed at codon 41 of the UBA1 gene, with the following frequencies: p.Met41Thr (c.122T > C), p.Met41Val (c.121A > G), and p.Met41Leu (c.121A > C), as observed in our patient4,5. The UBA1 gene encodes the ubiquitin-like modifier activating enzyme 1, a pivotal enzyme critical for cellular ubiquitylation. Ubiquitylation plays a crucial role in various cellular processes, including the regulation of the cell cycle, the response to DNA damage, and immune signaling pathways. Mutations in UBA1 result in diminished ubiquitination and activation of autoimmune pathways, leading to inflammatory manifestations5. These mutations result in reduced expression of the cytoplasmic UBA1b isoform and the formation of the catalytically impaired UBA1c isoform6. Although the p.Met41Leu mutation is associated with a more favorable prognosis, indicating higher residual UBA1b expression, our patient ultimately succumbed to disease-related complications6. Furthermore, it has been demonstrated that different genetic variants are linked to distinct clinical presentations. Patients with the Leu variant are more likely to be diagnosed with Sweet syndrome (as observed in our case), while patients with the Thr variants are more predisposed to developing inflammatory ocular diseases, and those with the Val variant are less likely to develop ear chondritis6.
Concerning cutaneous manifestations, these are frequently observed, with reports in up to 88% of patients, contingent on the patient cohorts4. The most prevalent cutaneous presentations may be non-vasculitic, including Sweet syndrome, neutrophilic dermatosis, erythema nodosum, urticaria, pressure plaques, injection site reactions, and others, or vasculitic, such as leukocytoclastic vasculitis3.
The UBA1 mutation was identified within skin tissues through genetic sequencing and analysis of samples obtained from VEXAS patients, mirroring the genetic anomalies seen in myeloid cells7. Consequently, it is theorized that the infiltration of dermal tissue arises from the clonal expansion of UBA1 mutant cells, rather than originating from an inflammatory context. In contrast, a separate study found that UBA1 mutations were exclusively detected in skin tissues of patients suffering from neutrophilic dermatosis and were notably absent in other dermatological conditions, such as leukocytoclastic cutaneous vasculitis8. This observation raises the possibility that UBA1 mutant clones may be absent in non-neutrophilic dermatitis or exist in quantities too small to be reliably detected. This disparity gives rise to different therapeutic strategies, either directed at targeting the UBA1 mutation or at ameliorating the inflammatory response3.
Given the systemic nature of VEXAS syndrome and its potential effects on multiple organ systems, including the lungs, blood vessels, and potentially the bone marrow, the application of computed tomography (CT) scanning arises as a valuable diagnostic tool. A CT scan provides detailed information regarding the extent of systemic involvement, especially in the thoracic region, which holds relevance for both precise diagnosis and the development of treatment strategies. This significance is emphasized by the patient’s pulmonary symptoms and the heightened incidence of pulmonary involvement observed in VEXAS syndrome3.
Patients may initially seek dermatological care for skin lesions, emphasizing the importance for dermatologists to consider VEXAS syndrome in their list of potential differential diagnoses. VEXAS syndrome should be contemplated in adult men exhibiting systemic symptoms, such as fever and fatigue, and presenting with sensitive, purpuric, or erythematous infiltrated nodules, periorbital edema, chondritis, or refractory vasculitis. Suspicion should be heightened if an abnormal blood count with cytopenias, elevated inflammatory markers, and a biopsy supporting neutrophilic dermatosis, with or without leukocytoclastic vasculitis, is present2. These patients should be referred for potential bone marrow examination to check for characteristic vacuoles in neutrophilic and erythroid precursors or for a definitive diagnosis through genetic sequencing.

