










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.34 no.3 Lisboa set. 2020
https://doi.org/10.32932/pjnh.2020.10.084
ORIGINAL ARTICLE
Kidney disease in tuberous sclerosis complex: A single-center experience
Joana Lorenzo1, Catarina Isabel Ribeiro2, Rita Russo Belo3, Liane Costa4, Ana Teixeira4, Liliana Rocha4, Teresa Costa4, Paula Pires Matos4, Maria do Sameiro Faria4, Conceição Mota4
1 Serviço de Pediatria, Centro Materno Infantil do Norte – Centro Hospitalar Universitário do Porto
2 Serviço de Nefrologia, Centro Hospitalar de Vila Nova de Gaia/Espinho
3 Serviço de Pediatria, Centro Hospitalar Trás-os-Montes e Alto Douro
4 Serviço de Nefrologia Pediátrica, Centro Materno Infantil do Norte – Centro Hospitalar Universitário do Porto
ABSTRACT
Introduction: Tuberous sclerosis complex is an autosomal dominant neurocutaneous disorder which is characterized by multisystem involvement, including the kidney. Since renal disease is one of the causes of morbidity and mortality, close management is essential. Subjects and Methods: In this retrospective observational,single‑center study, authors analyse patients with a definite diagnosis of tuberous sclerosis complex who attended Pediatric Nephrology consultation between 1998 and 2019. Results: 20 patients were included. The median age at the time of the diagnosis was 3 years, mostly after seizures and the median age of the first Pediatric Nephrology consultation was 9 years. Seventeen patients had renal disease, the majority with angiomyolipomas and a smaller number with cysts. None had hypertension or history of acute kidney failure. Systemic mTOR inhibitors were used in three patients. Discussion: The present results were similar to those found in the literature regarding presentation features and kidney disease. The authors emphasized the importance of early diagnosis of renal involvement in order to reduce renal morbidity and mortality (especially when these patients reach adulthood) and highlighted the need for early guidance for pediatric nephrology consultation.
Keywords: Kidney Disease, Pediatrics, Tuberous Sclerosis Complex
INTRODUCTION
Tuberous sclerosis complex (TSC) is an autosomal dominant neurocutaneous disorder caused by germline mutations in either the TSC1 or TSC2 genes, which encode hamartin and tuberin, respectively.1‑4
Genetic testing is often used in the diagnosis. It is a multisystemic disease with variable clinic expressiveness, and two‑thirds of the cases have de novo germline mutations. Approximately 70% of patients have TSC2 and present with a more severe disease compared to TSC1 mutations, which can be detected in about 20% of the cases.1,2 In the remaining 10%, no mutation can be identified by standard genetic sequencing, in some cases due to mosaicism. The estimated prevalence in Europe is 1/25,000 to 1/11,300.4
The characteristic features include multisystem hamartomas that affect the brain, kidney, eyes, heart, lung, liver and skin, and associated with neuropsychiatric features. For reasons not yet understood, disease severity and organ involvement vary widely between patients and within individuals of the same family who carry the same mutation.5
According to literature, clinical features of TSC have a distinct time of onset: rhabdomyomas can be detected in utero in the majority of patients; the dermatological features occur in childhood, and neurological findings with infantile spasms are also found in childhood.6
Renal disease is the important cause of morbidity and mortality in these patients. Since renal manifestations begin early during childhood, with an increased incidence and severity as the child ages, it is important to assess those manifestations regularly. The majority of patients develop multiple multifocal kidney angiomyolipomas; many have cysts, and a smaller number renal cell carcinoma, which can occur at an early age.
Renal damage may come from angiomyolipoma progression itself or from surgical and embolitic/ablative therapies, which are associated with increased risk of renal insufficiency and end‑stage renal failure.7,8
According to international guidelines for management and surveillance of TSC, mTOR inhibitors are the indicated drugs. Rapamycin was the first mTOR inhibitor studied for angiomyolipomas, but the largest randomized controlled trials that allowed the inclusion of mTOR in treatment protocols were about everolimus.9
SUBJECTS AND METHODS
The authors performed a retrospective observational study which included patients with a definite TSC diagnosis who attended a Pediatric Nephrology consultations during the last twenty‑two years.
Twenty patients were eligible for inclusion and data were collected in an anonymized database.
Data on demographics, age and presentation feature at time of the diagnosis and organ involvement were taken from patients’ electronic medical records. Last available kidney function, the presence of hypertension and the presence of angiomyolipomas and cysts were recorded from the last available reports, as well as the treatment used. Finally, other comorbidities were recorded, namely cardiac, neurological and cutaneous lesions.
Statistical analysis was performed using IBM SPSS Statistics®, version26. For the descriptive analysis, means and standard deviations of continuous variables with symmetry of distributions were presented; in the case of asymmetric distributions, the median was used, mentioning the minimum and maximum values.
RESULTS
Twenty patients were included, male gender 600% (n=12). The median age at diagnosis was 3 [0.2‑13.2] years. There was a family history of TSC in 3 patients (15%). Eighteen patients (90.0%) were symptomatic at diagnosis, with the presenting feature being seizures in 16 cases (88.8%) and hypochromic skin lesions in 2 patients (11.2%). In the asymptomatic patients, one was diagnosed in the context of a positive family history of TSC, and the other case a cardiac rhabdomyoma was diagnosed during the neonatal period due to dependence on oxygen.
Six patients had TSC2 mutations and two had TSC1 mutations. In terms of comorbidities, all patients had neurological manifestations; seventeen patients had epilepsy (85.0%), 7 (35.0%) had ubependymal giant cell astrocytomas (SEGA), 6 (30.0%) had tubers and 4 (20.0%) had subependymal nodules. Fourteen patients (70.0%) had cutaneous manifestations, with angiofibromas in 9 (64.3%), Shagreen patches in 1 (7.1%) and 4 (28.6%) having both. Cardiac rhabdomyomas were found in 12 patients (60.0%). Data regarding patients’ characteristics are described in Table I.
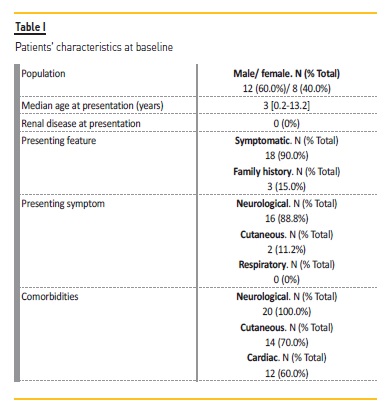
The first Pediatric Nephrology consultation was at 9 [1.5‑17.8] years.
Renal manifestations were found in 17 patients (85.0%), with angiomyolipomas found in 15 (75.0%) and cysts in 9 (45.0%). Kidney function was available for 16 patients (80.0%); the mean serum creatinine levels at the time of the last consultation were 0.58 mg/dL. No patient had episodes of AKI. Through follow‑up, none developed stage 3 or more chronic kidney disease according to Kidney Disease Improving Global Outcomes (KDIGO) or have been submitted to kidney transplant.
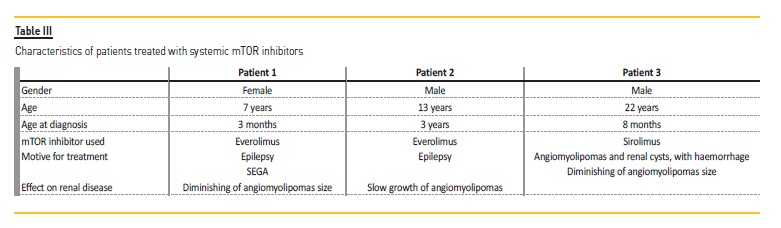
No patient had arterial hypertension. In the 17 patients with renal manifestations (described in Table II), nine were treated with topical mTOR inhibitors due to facial angiofibromas. Systemic mTOR inhibitors were used in three patients, with everolimus being used in 2 cases and sirolimus in one. All three patients had renal manifestations of the disease, having started the therapeutics due to epilepsy in two cases.
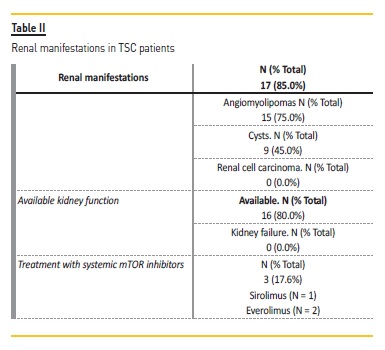
In Table III there is a description of the 3 patients treated with systemic mTOR inhibitors.
Almost all patients had follow‑up consultations (n=19), with 2 patients being followed by Nephrology consultation, since they have reached adulthood. The median time of follow‑up was 2.6 [0.6‑20.6] years. Only one patient (Patient 3) showed hemorrhagic angiomyolipomas; since it was possible to reduce angiomyolipomas’ size and control the bleeding, neither embolization or nephrectomy were performed.
No patient died during this time. Thirteen patients (65.0%) were followed by psychologist and/or psychiatrist.
DISCUSSION
Numbers show nearly 1500 persons with TSC in Portugal and one million around the world.10 In this twenty‑two‑year study, the authors found 20 patients with TSC followed by Pediatric Nephrology consultation, a significant number according to the national panorama.
TSC is an underdiagnosed disease in many cases due to disease complexity and different forms of presentation.6 In our sample, the majority of patients were symptomatic at the time of diagnosis, with seizures as the main feature, as shown in literature. Only 3 cases had a positive family history, and eight had TSC1 or TSC2 mutations, which is in agreement with the high rate of de novo mutations characteristicof this disease.
Dermatological changes are present in the majority of patients.11
In the present sample, cutaneous manifestations were found in 70% of the patients, numbers similar to those found in literature. Treatment with topical mTOR inhibitors in more than half of the patients with cutaneous findings may be a reflection of concerns of systemic mTOR inhibitors due to their systemic side effects and due to a subjective better quality of life when using the topical drugs.
In terms of renal disease, multiple renal angiomyolipomas are frequent, with an estimated incidence of 55 to 75%.12 Cysts are also a common lesion, generally asymptomatic and associated with hypertension and renal failure.13 Although none of the patients presented with hypertension, this is a modifiable progression factor that occurs frequently and early in TSC patients, that should have a preventive management. In fact, hypertension and also decline in glomerular filtration rate may occur in TSC patients, but didn’t happen in our study. A possible explanation may be the avoidance of more invasive procedures (such as nephrectomy or embolization), that would be an important factor in increased risk of renal failure; further, the possible renal damage due to mTOR inhibitors was reduced, as all patients had normal renal function prior to drug initiation.9 As recommended by international guidelines for TSC, mTOR inhibitors should be used in growing angiomyolipomas larger than 3 cm in diameter, with a positive effect in reducing their size as shown in Patient 3 of our study.10
Due the multisystemic character of the disease, it requires a multidisciplinary approach, with coordination of all health professionals.
According to international guidelines, kidney and brain imaging (abdominal and brain magnetic resonance imaging, respectively), blood pressure measurement and kidney function evaluation, dermatologic exam and evaluation for evidence of neuropsychiatric/neurodevelopmental disorders should be repeated at least annually.10
It is important to be aware of the possible side effects of mTOR inhibitors such as mouth ulcers, acne‑like skin lesions, nasopharyngitis, hyperlipidemia and peripheral edema, as well as possible renal damage as described before.
This study has some limitations, which includes the small number of patients and the difficulty in accessing all the important data, since many patients had their clinical files on paper.
References
1. Dabora SL et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001; 68: 64–80 [ Links ]
2. Henske EP et al. Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13 occurs in sporadic as well as TSC‑associated renal angiomyolipomas. Genes Chromosom Cancer 1995; 13: 295–298 [ Links ]
3. Van Slegtenhorst M et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277: 805–808 [ Links ]
4. ORPHANET [homepage na Internet]. Esclerose Tuberosa [acesso em dezembro de 2019]. Disponível em https://www.orpha.net [ Links ]
5. Lam HC, Siroky BJ, Henske EP. Renal disease in tuberous sclerosis complex: pathogenesis and therapy. Nat Rev Nephrol; 2018; 14: 704–716 [ Links ]
6. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006; 355: 1345–1356 [ Links ]
7. Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin. Proc. 1991; 66: 792–796 [ Links ]
8. Eijkemans MJ et al. Long‑term follow‑up assessing renal angiomyolipoma treatment patterns, morbidity, and mortality: an observational study in tuberous sclerosis complex patients in the Netherlands. Am. J. Kidney. Dis. 2015; 66: 638–645 [ Links ]
9. Amin S et al. The UK guidelines for management and surveillance of Tuberous Sclerosis Complex. QJM. 2019 Mar 1;112(3):171‑182 [ Links ]
10. Associação de Esclerose Tuberosa em Portugal [homepage in the Internet; access in December 2019]. Disponível em: http://www.esclerosetuberosa.org.pt [ Links ]
11. Diagnóstico e Seguimento da Pessoa com Esclerose Tuberosa em Idade Pediátrica e no Adulto. Norma de Orientação Clínica da Direcao‑Geral da Saúde. 2018 [ Links ]
12. Ewalt DH, Sheffield E, Sparagana SP, Delgado MR, Roach ES. Renal lesion growth in children with tuberous sclerosis complex. J Urol 1998; 160: 141–145 [ Links ]
13. O’Callaghan FJ, Noakes MJ, Martyn CN, Osborne JP. An epidemiological study of renal pathology in tuberous sclerosis complex. BJU Int 2004; 94: 853–857
Joana Lorenzo
Centro Materno Infantil do Norte – Centro Hospitalar Universitário do Porto
Largo da Maternidade Júlio Dinis, 4050‑371
Porto, Portugal
E‑mail: joanalorenzof@gmail.com
Disclosure of potential conflicts of interest: none declared
Received for publication: Jul 15, 2020
Accepted in revised form: Sep 23, 2020


{kind=link}