











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Eosinophilic granulomatosis with polyangiitis (EGPA) is a systemic small and medium sized vessel vasculitis.1 Its presentation is usually divided into three phases: prodromal, eosinophilic and vasculitic.2
Life-threatening complications normally take place in the last phase.3 This syndrome affects predominantly the respiratory system, heart, skin and peripheral nerves.4 Renal involvement occurs in about one quarter of the cases and the glomerulus is usually the target, exhibiting a necrotizing crescentic glomerulonephritis pattern and presenting
clinically as a rapidly progressive kidney failure.4,5However, there have been reports of rare cases of acute interstitial nephritis with or without glomerular lesions.6,7EGPA diagnosis is usually suspected in a patient with history of asthma and rhinosinusitis
along with the identification of peripheral eosinophilia.1,3,8Positivity of p-ANCA is not a sensitive diagnostic tool, but it is noted in approximately 30% of cases.4,9The diagnosis can be corroborated with biopsy from affected tissues, such as respiratory mucosa, skin and peripheral nerves.1,10,11 Treatment consists of systemic corticosteroids and other immunosuppressors, such as cyclophosphamide and azathioprine.1,12,13Prognosis improves greatly after introduction of immunosuppressive therapy, reaching a 5-year survival rate around 80%.14,15
CASE REPORT
We present the case of a 60-year-old caucasian man with a personal background that included asthma, rhinosinusitis and a stroke in the previous year, leaving him with right hemiparesis as sequelae. His medication comprised mometasone furoate, an inhaler (budesonide plus formoterol), pantoprazole, carvedilol and alprazolam.
The patient presented to the emergency room with painless and sudden loss of visual acuity along with a purpuric exanthem in his lower limbs that had surged 24 hours before. He also reported astenia and myalgia in the last two weeks. Head CT scan ruled out stroke and intracranial lesions. Further examination discriminated the impairment in his visual acuity (VA): left eye (LE) could only see 2/10 and his right eye (RE) was only slightly affected (VA 8/10). Optical coherence tomography (OCT), performed 24 hours after the episode, showed evidence of left central retinal artery occlusion and perfusion deficits in the superior portion of the right arterial retinal blood supply. No treatment was employed at this time. The patient returned to the emergency department five days later referring worsening of his visual impairment.
This time he could only see shadows from his left eye and his right eye’s VA had also worsened (6/10). Complementary study (Table I) showed prominent peripheral eosinophilia (21.800 cel/uL; 64% of all leukocytes), increased serum IgE (1260U/L) and increased C-reactive protein (10.6mg/dl). His kidney function was normal. The patient was admitted and, four days after admission, presented with acute kidney failure, exhibiting a serum creatinine (sCr) of 1.7mg/dl. Until that time, the patient had only been prescribed with his usual medication, paracetamol and venous thromboembolism prophylaxis. Further workup revealed exceptionally high p-ANCA MPO titer (>600U/L).
Other causes of hypereosinophilic syndrome were excluded. A bronchoalveolar lavage and bronchial biopsy were performed and both revealed intense eosinophilic infiltrate. Skin biopsy also revealed high eosinophil content as well as small vessel vasculitis. Kidney function kept worsening and, seven days after admission, sCr reached
4.7mg/dl (Figure 1).
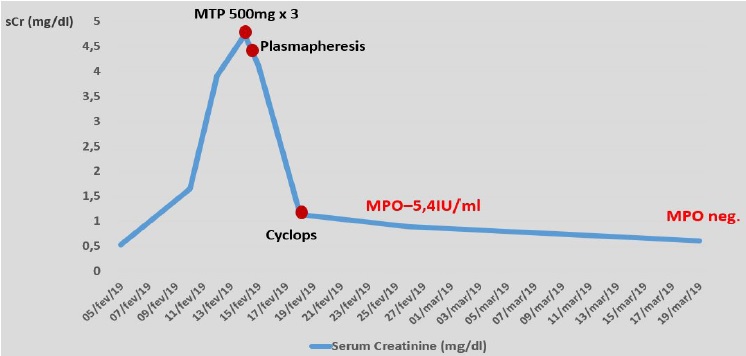
Figure 1 Patient’s renal function (serum creatinine) evolution from hospital admission until serum ANCA-MPO turned negative, as well as its relation to therapeutic approach.
Urinary sediment showed signs of inflammatory activity with proteinuria and microscopic hematuria (22 erythrocytes/HPF).
Considering the acute kidney injury, prominent eosinophilia and strongly positive anti-MPO antibodies, EGPA was diagnosed and pulse steroid therapy was administered, followed by intravenous cyclophosphamide (cyclops protocol). Plasmapheresis was also provided (seven sessions), since the treatment was started before kidney biopsy results became available. Kidney biopsy (figure 2) showed acute tubulointerstitial nephritis with prominent eosinophil infiltrate and only mild glomerular involvement. His kidney function improved promptly and one week later it had already reached normal values. Renal replacement therapy was not required. Patient’s purpuric exanthem was completely resolved. However, VA defects did not improve.
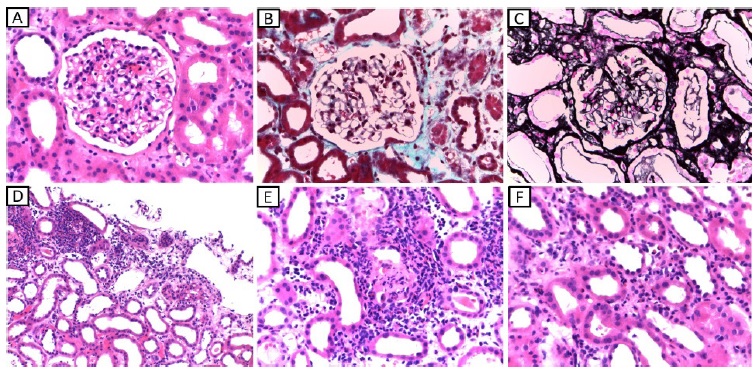
Figure 2 A - light microscopy (HE, 400x) showing glomerulus with normal morphology with only light endocapillary cellularity (neutrophils and lymphocytes). B - light microscopy (Masson’s trichrome stain, 400x) showing glomerulus with properly thin capillary walls and without thrombi. C - light microscopy (Marinozzi, 400x) showing glomerulus with thin capillary walls, without sclerosis, spikes or double contours. D - light microscopy (HE, 100x) showing tubular atrophy accompanied by edema, mild interstitial fibrosis and areas of intense inflammatory infiltrate at the expense of neutrophils, lymphocytes and eosinophils which permeate the tubular epithelium causing tubulitis. E - light microscopy (HE, 200x) showing inflammatory infiltrate at the expense of lymphocytes, neutrophils and eosinophils with permeation of the tubular epithelium causing destruction of the epithelium and formation of granular cylinder. F - light microscopy (HE, 400x) showing the presence of inflammatory cells (lymphocytes, neutrophils and eosinophils) in the lumen of intertubular capillaries (capillaritis-type lesions).
DISCUSSION
Eosinophilic granulomatosis with polyangiitis is the least common of the ANCA-associated vasculitis. It is also the one which most often spares the kidneys, targeting mainly the lungs and skin.1,9It is unusual in individuals over 65 years old, having a mean age at diagnosis of 40 years.4 Patients usually have a background of asthma and chronic rhinosinusitis.16 Although we had no written reports, our patient had already been medicated with inhaled corticosteroids for 6 years, assuming a history of bronchial hyperreactivity.
He was also prescribed mometasone furoate, presumably for rhinosinusitis. Further, the head CT scan showed total opacification of the paranasal sinuses.
ANCA antibodies are found in about half the cases, mainly p-ANCA.4,19 Other laboratory findings include normocytic and normochromic anemia, eosinophilia, elevated inflammatory markers, hypergammaglobulinemia with an elevated serum immunoglobulin E, and positive rheumatoid factor.3,20,21Our patient had positive p-ANCA with a very high titer (>600UI/ml), a pronounced eosinophilia, a high immunoglobulin E titer and elevation of inflammatory markers. These analytical findings, along with the biopsy samples obtained, were in agreement with the diagnosis of EGPA and allowed us to relate each tissue damage to this syndrome. We confirmed the diagnosis of EGPA using the American College of Rheumatology criteria, which requires the presence of four or more criteria (asthma; eosinophilia > 10%; mono-or polyneuropathy attributable to systemic vasculitis; pulmonary infiltrates; paranasal sinusitis and biopsy demonstrating extravascular accumulation of eosinophils) in order to diagnose this disorder with a sensitivity of 85 percent and a specificity of 99.7 percent.22 In the case presented, four of these criteria were met.
Hypereosinophilic syndrome, as well as other secondary causes of hypereosinophilia, were excluded. Dress syndrome was discarded, considering that a suspicious high-risk drug was not identified, nor did the patient present with fever, enlarged lymph nodes or a skin rash suggestive of the disease. Parasite cultures and serologies for parasites were negative. Serum immunophenotyping, FIP1L1-PDGFRA fusion gene test and BCR-ABL 1 t(9;22)(q34;q11.2) test (FISH technique) were negative. Furthermore, the patient had no clinical or serological evidence of active Epstein-Barr infection.
None of the other ANCA-associated vasculitis would have had such a clinical presentation, particularly the prominent eosinophilia.
Other causes of acute kidney injury were also excluded. Although one may argue the patient was more likely to have an acute interstitial nephritis secondary to a drug administered during hospitalization, no culprit drug was identified. At the time the acute kidney failure was diagnosed, the patient was medicated with low molecular weight heparin, aspirin, carvedilol, escitalopram, paracetamol, atorvastatin, loratadine, pantoprazole and ipratropium bromide.
The patient had already been chronically medicated with most of these drugs. None of these were withdrawn, in agreement with our clinical suspicion, and the patient’s kidney function improved and remained stable thereafter.
Eosinophil tissue infiltration and necrotizing granulomas are the major histologic findings as the syndrome name implies. However, the absence of granulomas in biopsy samples is not unusual.17 The kidney is only affected in 25% of cases.4,5It happens most frequently through a rapidly progressive necrotizing crescentic glomerulonephritis.
However, there have been case series reports on EGPA patients who underwent kidney biopsy showing rare cases of eosinophilic interstitial nephritis (around 15%), with or without glomerular involvement. 6,7In both of these series, among a total of 35 kidney biopsies, only two (5,7%) showed eosinophilic interstitial nephritis as the dominant
feature.6,7Kidney biopsy from our patient showed only mild endocapillary proliferation, the predominant feature being eosinophilic interstitial nephritis.
The patient’s visual loss was thought to be due to the systemic vasculitis, considering its presentation timing. Head CT scan excluded any major acute vascular accident or mass effect with nerve compression. OCT showed evidence of left central retinal artery occlusion. This is, fortunately, an exceptionally rare occurrence in this disorder. It is usually irreversible and thought to be due to anterior ischemic optic neuropathy precipitated by vascular inflammation.23,24,25,26,27EGPA treatment consists of systemic corticosteroids and other steroid-sparing immunosuppressors. Since the patient had evidence of acute multiorgan damage, three methylprednisolone pulses (500mg) and intravenous cyclophosphamide (cyclops protocol) were provided. Plasmapheresis is normally reserved for patients with rapidly progressive glomerulonephritis and diffuse alveolar hemorrhage. Since treatment was readily started, before kidney biopsy results were available, plasmapheresis was provided, considering EGPA kidney involvement is most frequently the result of a necrotizing crescentic glomerulonephritis.
Prognosis has improved much after the introduction of immunosuppressive arsenal, with a 5-year survival rate around 80%.14,15
Our patient’s acute kidney failure completely resolved after the initiation of corticosteroid and cyclophosphamide therapy. At discharge, his kidney function was already normal. This exceptionally good response was probably due to the fact that the patient had only evidence of interstitial nephritis, without significant glomerular involvement. After the full cyclops protocol (10 pulses of cyclophosphamide), azathioprine therapy (1.5mg/kg/day) was provided. In the meantime, corticosteroid therapy was tapered according to the pexivas protocol, as would have been done with any other ANCAassociated vasculitis with a resembling clinical picture. Currently, after one-year follow-up, the patient has a normal kidney function, a benign urinary sediment and undetectable p-ANCA antibody titer.
He has been on azathioprine for six months. Unfortunately, his visual deficit had not improved significantly (VA-LE: 1/10; VA-RE:8/10) at his last ophthalmology appointment.
This case describes a curious and dramatic EGPA clinical presentation along with an unusual kidney involvement that responded well to the immunosuppressive therapy, contributing to the relatively limited nephrological experience in this syndrome.18 28,29,30,31,32

