










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Medicina Geral e Familiar
versão impressa ISSN 2182-5173
Rev Port Med Geral Fam vol.36 no.5 Lisboa out. 2020
https://doi.org/10.32385/rpmgf.v36i5.12567
RELATOS DE CASO
Microcefalia: um caso de fenilcetonúria materna não diagnosticada
Microcephaly: a clinical case of maternal phenylketonuria undiagnosed
Mariana Abreu,1
 https://orcid.org/0000-0002-6743-3068
https://orcid.org/0000-0002-6743-3068
Joana Jardim,2
João Luís Barreira3
1 Médica Interna de Pediatria. Centro Hospitalar e Universitário de São João.
2 Médica Assistente de Pediatria. Hospital Pediátrico Integrado, Centro Hospitalar e Universitário de São João.
3 Médico Assistente Graduado de Pediatria. Hospital Pediátrico Integrado, Centro Hospitalar e Universitário de São João.
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Introdução: A microcefalia e a microencefalia que lhe está associada pode ter início intrauterino ou pós-natal. A etiologia é diversa, englobando doenças genéticas, metabólicas, infeções intrauterinas ou exposições ambientais, exigindo uma abordagem diagnóstica específica.
Descrição do caso: Apresenta-se uma lactente de 10 meses, com história de restrição do crescimento intrauterino, microcefalia, atraso do desenvolvimento psicomotor e dismorfias faciais. A mãe evidenciava atraso intelectual e tinha outro filho com microcefalia e atraso do desenvolvimento psicomotor. Suspeitou-se de um fator materno na génese deste quadro. Os níveis séricos maternos de fenilalanina foram compatíveis com o diagnóstico de fenilcetonúria clássica, identificando-se a síndroma de fenilcetonúria materna.
Comentário: Níveis séricos elevados de fenilalanina durante a gravidez são teratogénicos para o feto e associam-se a restrição do crescimento intrauterino, microcefalia, cardiopatias congénitas, dismorfias faciais, atraso de crescimento pós-natal e atraso do desenvolvimento psicomotor. Esta embriopatia pode ser prevenida pela restrição dietética de fenilalanina, iniciada antes da conceção e mantida na gravidez.
Palavras-chave: Microcefalia; Hiperfenilalaninemia; Fenilcetonúria, Clássica; Embriopatias; Fenilcetonúria, Materna.
ABSTRACT
Introduction: Microcephaly is associated with microencephaly and it may be present at birth or may develop in the postnatal period. The pathogenesis of microcephaly is heterogeneous, ranging from genetic causes, metabolic diseases, intrauterine infections, or environmental factors. A specific approach is needed.
Clinical case: We present a 10 months old female with intrauterine growth restriction, microcephaly, psychomotor developmental delay, and facial dysmorphism. Her mother revealed intellectual disability and had another son with microcephaly and psychomotor developmental delay. We suspected a maternal condition to explain this. Maternal serum levels of phenylalanine were consistent with classic phenylketonuria, placing the diagnosis of maternal phenylketonuria syndrome.
Discussion: Elevated phenylalanine levels in pregnant women are teratogenic and can result in intrauterine growth restriction, microcephaly, cardiac malformations, facial dysmorphism, delayed postnatal growth, and psychomotor developmental delay. This embryopathy can be prevented by dietary restriction of phenylalanine before and during pregnancy.
Keywords: Microcephaly; Hyperphenylalaninaemia; Phenylketonuria, Classical; Embryopathies; Phenylketonuria, Maternal.
Introdução
O perímetro cefálico relaciona-se com o crescimento do sistema nervoso central. A microcefalia define-se como perímetro cefálico dois desvios-padrão abaixo da média para a idade e o sexo.1 A microcefalia pode estar presente ao nascimento ou desenvolver-se posteriormente, evidenciando-se habitualmente nos dois primeiros anos de vida, quando o crescimento cerebral é acelerado. As crianças afetadas têm frequentemente atraso do desenvolvimento psicomotor.2
A microcefalia pode ser a manifestação inicial de diversas patologias. Pode ser desencadeada por alterações cromossómicas, genéticas (e.g., erros inatos do metabolismo), infeções intrauterinas (e.g., rubéola, citomegalovírus ou toxoplasmose), exposição pré-natal a tóxicos e doenças endócrinas ou metabólicas maternas (e.g., hipotiroidismo e hiperfenilalaninemia).2-4
Perante uma criança com microcefalia é essencial uma história clínica detalhada, com dados pré-natais, do parto, antecedentes patológicos, desenvolvimento psicomotor e antecedentes familiares. É importante documentar o perímetro cefálico dos pais, porque pode representar um traço hereditário.2 Exige um exame objetivo completo, avaliando características dismórficas, alterações neurológicas e avaliação da presença de atraso do desenvolvimento psicomotor.2-3
A investigação subsequente depende da situação ser sindrómica ou isolada e se teve início pré ou pós-natal.1-4 A ressonância magnética cerebral é o exame de escolha, identificando anomalias estruturais, transtornos da migração neuronal e defeitos da mielinização. Muitas das condições associadas têm fenótipos específicos, permitindo testes direcionados.1-5
Descrição do caso
Apresenta-se uma lactente com 10 meses de idade, encaminhada pelo médico assistente para a consulta de pediatria geral por microcefalia. A sua gestação foi vigiada, sendo a mãe imune para a toxoplasmose e rubéola no primeiro trimestre; no terceiro trimestre as serologias para VDRL, vírus de imunodeficiência humana e hepatite B eram negativas. Na ecografia do segundo trimestre foi registada restrição do crescimento intrauterino (RCIU) grave, tendo efetuado amniocentese, com cariótipo 46, XX. Foram excluídas outras infeções durante a gestação, exposição a radiações, consumo de álcool, drogas ou outros fármacos para além dos suplementos de ácido fólico e ferro. O parto ocorreu neste hospital às 34 semanas por cesariana eletiva (por RCIU), necessitando de reanimação com ventilação com pressão positiva. Índice de Apgar 1º/5º minutos de 7/9. Antropometria ao nascimento com peso de 1485g, comprimento 38cm e perímetro cefálico 26cm, todos abaixo do percentil (P) 3 nas Curvas de Fenton. Teve internamento por prematuridade e baixo peso em hospital de proximidade sem intercorrências assinaladas. Acompanhamento médico posterior irregular por falta de comparência a consultas. Registo de evolução estaturo-ponderal nos primeiros meses de vida sempre abaixo do P5, com perímetro cefálico muito inferior ao P5 para a idade cronológica corrigida.
A mãe tinha 32 anos e apresentava aparente perturbação intelectual. Tinha história obstétrica de três gestações, com uma interrupção voluntária de gravidez às cinco semanas. Negava hábitos alcoólicos ou tabágicos. O pai, de 52 anos, não alegava antecedentes patológicos relevantes. Sem consanguinidade. Meio-irmão da parte materna, com sete anos, aos cuidados do pai, acompanhado em consulta multidisciplinar por apresentar uma síndroma dismórfica não esclarecida, com microcefalia e atraso global do desenvolvimento. Tinha três meios-irmãos por parte paterna, de 34, 33 e 19 anos, aparentemente saudáveis (Figura 1).
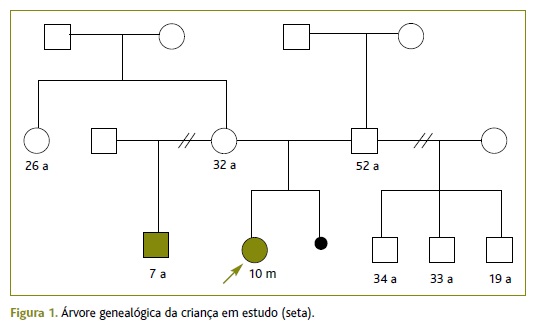
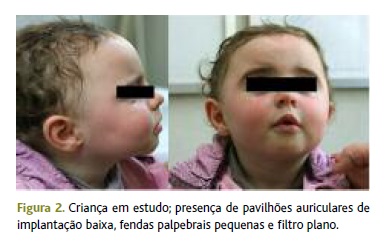
Na observação inicial na consulta, aos 10 meses, foi objetivada uma fácies peculiar Figura 2) e atraso global do desenvolvimento, sobretudo ao nível da motricidade grosseira (não se sentava sem apoio nem rastejava).
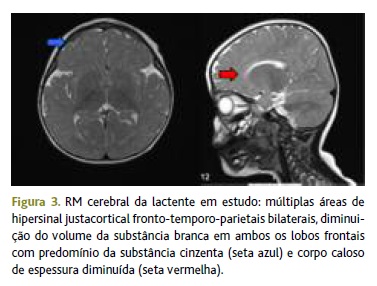
O estudo analítico efetuado (hemograma, ionograma, testes hepáticos e renais, amónia e lactato), bem como o estudo ecográfico abdominal e reno-pélvico não revelaram alterações. Foi avaliada por oftalmologia e cardiologia, tendo sido excluída patologia ocular ou cardíaca. A ressonância magnética cerebral demonstrou alterações inespecíficas (Figura 3).
Os antecedentes familiares desta criança, nomeadamente a existência de um meio-irmão materno, também com microcefalia e atraso do desenvolvimento psicomotor, fizeram suspeitar da presença de um fator materno, comum às duas gestações, como desencadeante da patologia da doente em análise. A investigação concentrou-se na aparente perturbação intelectual materna, tendo-se proposto colheita de sangue materno em cartão de Guthrie para doseamento de fenilalanina (Phe) sérica. Foi registado um valor de Phe de 1215µmol/L, muito elevado, compatível com o diagnóstico de fenilcetonúria (PKU) clássica. Este resultado foi confirmado numa segunda determinação.
Comentário
Pensando numa causa gestacional para este quadro, dever-se-ia equacionar uma doença materna genética, endócrina ou metabólica. Outra hipótese seria a exposição a um agente teratogénico (e.g., álcool), que teria de se ter mantido nas duas gestações. Contudo, foram negadas exposições de risco. O estudo efetuado foi direcionado para a perturbação intelectual materna.
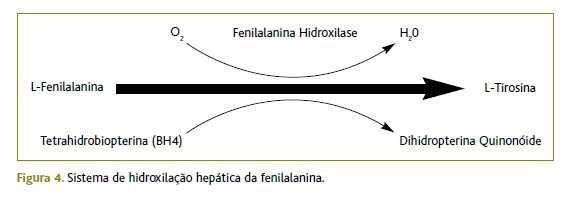
A PKU é a patologia congénita mais comum do metabolismo dos aminoácidos.6-8 Ocorre em todos os grupos étnicos e a incidência pode variar de 1:2.600 a 1:26.000, sendo a média de 1:10.000.9-11 A incidência desta doença em Portugal é de 1:10.772 recém-nascidos, mantendo-se estável ao longo dos anos.7,12 É uma doença de transmissão autossómica recessiva, desencadeada por mutações num gene localizado no cromossoma 12 (12q22-24), que codifica a enzima hepática fenilalanina-hidroxilase (FH). Essa enzima é responsável pela utilização de 75% da Phe da dieta. A tetrahidrobiopterina (BH4) é um co-fator necessário à atividade da FH. Os defeitos no metabolismo da BH4 são responsáveis por 2% dos casos de doença. A ausência ou deficiência da FH impede a conversão hepática de Phe, um aminoácido essencial, em tirosina (Figura 4). Ocorre acumulação de Phe e dos seus precursores e a tirosina torna-se um aminoácido essencial.6,9,13
O defeito metabólico conduz à redução da síntese de uma grande variedade de neurotransmissores, sendo a serotonina o mais deficitário. Pensa-se que o excesso de Phe interfere no desenvolvimento e na mielinização do sistema nervoso.7,9,11,13-14
O défice completo da FH causa PKU Clássica (Phe sérica > 1200µmol/L), valor constatado na mãe da criança em estudo. Uma atividade enzimática parcial condiciona PKU Moderada (Phe sérica 600-1200µmol/L) ou hiperfenilalaninemia (Phe sérica 150-600µmol/L).10,13-15 O controlo metabólico pode ser alcançado através de uma dieta rigorosa, isenta de Phe em combinação com suplementos de tirosina.9,14,16-17 A sapropterina, uma forma sintética da BH4, demonstrou ser eficaz na redução dos níveis de Phe em alguns pacientes com PKU, quando usada como adjuvante da restrição dietética, permitindo uma dieta mais liberal.14,18-19
O rastreio neonatal universal desta patologia fez com que as manifestações clínicas típicas desta doença sejam atualmente raramente encontradas. Se o defeito enzimático não for detetado pelo rastreio neonatal, a evolução da doença é insidiosa. Nos doentes não tratados, a principal manifestação é a perturbação intelectual,14 evidente na mãe da doente apresentada. Como após a adolescência os efeitos indesejados do consumo de Phe são mínimos, muitas mulheres com doença interrompem a dieta.9,14,16-17 Essas mulheres podem ter um desenvolvimento cognitivo normal, fruto da intervenção dietética precoce.17
Numa gestação, a Phe atravessa a placenta resultando numa concentração fetal 70-80% superior à concentração materna. Níveis séricos maternos elevados de Phe (sobretudo > 360µmol/L) são teratogénicos, conduzindo a uma embriopatia denominada síndroma de PKU materna. Esta síndroma caracteriza-se por RCIU, microcefalia, atraso global do desenvolvimento, cardiopatias congénitas e dismorfias faciais.13,15,23 A criança em análise apresentava todos estes problemas, exceto cardiopatia. A ocorrência de microcefalia e de perturbações do desenvolvimento é significativamente mais frequente nos filhos de mães doentes. As cardiopatias congénitas e as dismorfias faciais são também encontradas nestas crianças, mas em menor percentagem. As cardiopatias ocorrem geralmente quando os valores de Phe sérica são muito altos e a sua ocorrência diminui significativamente se houver cumprimento da dieta durante a gestação, independentemente dos níveis maternos de défice enzimático.17,23 Níveis elevados de Phe não foram associados a aumento de complicações da gravidez, nomeadamente não está descrito aumento das taxas de morte fetal ou de partos pré-termo.17-22
A dieta deve ser rigorosamente estabelecida, idealmente antes da conceção ou durante as primeiras semanas do primeiro trimestre da gestação.13-17,19,23 Os efeitos deletérios sobre o feto diminuem quando os níveis de Phe do plasma da mãe são mantidos entre 120-360µmol/L.10,19
O rastreio neonatal da PKU iniciou-se em Portugal em 1979 através do Programa Nacional para o Diagnóstico Precoce. Mulheres que tenham falhado o rastreio podem ter esta doença não diagnosticada.7-8 A mãe desta doente nasceu no domicílio e a falta de contacto com os serviços médicos terá sido o motivo para não ter realizado o rastreio neonatal. Assim, apesar de ter nascido após a implementação do rastreio no nosso país, só teve diagnóstico de PKU Clássica após ter tido dois filhos que, apesar de potencialmente saudáveis, sofreram os efeitos teratogénicos da doença materna. O seguimento médico inadequado da mãe, que determinou a ausência de diagnóstico da sua doença e o facto do primeiro filho se encontrar sob a guarda exclusiva do pai, terão sido fatores desfavoráveis no estudo etiológico. Estamos perante o caso de três doentes com sequelas irreversíveis, mas todas elas atualmente preveníveis.
Sugere-se a implementação de medidas de determinação dos níveis séricos de Phe em mulheres em idade reprodutiva que apresentem perturbação do desenvolvimento intelectual. É também essencial manter o seguimento das jovens com doença conhecida, alertando-as para a necessidade de um controlo metabólico rigoroso antes e durante a gravidez.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Ashwal S, Michelson D, Plawner L, Dobyns WB. Practice parameter: evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2009;73(11):887-97. [ Links ]
2. Wroblewski ME, Bevington J, Badik C. Head growth. Pediatr Rev. 2015;36(9):426-7. [ Links ]
3. Boom JA. Microcephaly in infants and children: etiology and evaluation. UpToDate [Internet]; 2019 [updated 2019 Jan 21; cited 2019 Jun 29]. Available from: https://www.uptodate.com/contents/microcephaly-in-infants-and-children-etiology-and-evaluation [ Links ]
4. Von der Hagen M, Pivarcsi M, Liebe J, von Bernuth H, Didonato N, Hennermann JB, et al. Diagnostic approach to microcephaly in childhood: a two-center study and review of the literature. Dev Med Child Neurol. 2014;56(8):732-41. [ Links ]
5. Abuelo D. Microcephaly syndromes. Semin Pediatr Neurol. 2007;14(3):118-27. [ Links ]
6. Van Spronsen FJ. Phenylketonuria: a 21st century perspective. Nat Rev Endocrinol. 2010;6(9):509-14. [ Links ]
7. Vilarinho L, Queirós A, Leandro P, Almeida IT, Rivera I. Fenilcetonúria revisitada [Phenylketonuria revisited]. Arq Med. 2006;20(5-6):161-72. Portuguese
8. Osório RV, Vilarinho L, Soares JP. Rastreio nacional da fenilcetonúria, hipotiroidismo congénito e hiperplasia congénita das suprarrenais [National screening for phenylketonuria, congenital hypothyroidism and congenital adrenal hyperplasia]. Acta Med Port. 1992;5(3):131-4. Portuguese
9. Feldmann R, Osterloh J, Onon S, Fromm J, Rutsch F, Weglage J. Neurocognitive functioning in adults with phenylketonuria: report of a 10-year follow-up. Mol Genet Metab. 2019;126(3):246-9. [ Links ]
10. Mira NV, Marquez UM. Importância do diagnóstico e tratamento da fenilcetonúria [Diagnoses and treatment of phenylketonuria]. Rev Saúde Públ. 2000;34(1):86-96. Portuguese
11. Di Meglio G. Phenylketonuria. Pediatr Rev. 1998;19(6):214-5. [ Links ]
12. Vilarinho L, Garcia P, Costa PP. Programa nacional de diagnóstico precoce: relatório 2017. Lisboa: Instituto Nacional de Saúde Doutor Ricardo Jorge; 2018. [ Links ] ISBN 9789898794543
13. Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188-200. [ Links ]
14. Bodamer OA. Overview of phenylketonuria. UpToDate [Internent]; 2018 [updated 2018 Dec 3; cited 2019 Jun 29]. Available from: https://www.uptodate.com/contents/overview-of-phenylketonuria [ Links ]
15. Committee on Genetics. Maternal phenylketonuria. Pediatrics. 2008;122(2):445-9. [ Links ]
16. Koch R, Trefz F, Waisbren S. Psychosocial issues and outcomes in maternal PKU. Mol Genet Metab. 2010;99 Suppl 1:S68-74. [ Links ]
17. Prick BW, Hop WC, Duvekot JJ. Maternal phenylketonuria and hyperphenylalaninemia in pregnancy: pregnancy complications and neonatal sequelae in untreated and treated pregnancies. Am J Clin Nutr. 2012;95(2):374-82. [ Links ]
18. Eshraghi P, Asl SN, Bagheri S, Chalak V. Response to sapropterin hydrochloride (Kuvan®) in children with phenylketonuria (PKU): a clinical trial. J Pediatr Endocrinol Metab. 2019;32(8):885-8.
19. Manta-Vogli PD, Dotsikas Y, Loukas YL, Schulpis KH. The phenylketonuria patient: a recent dietetic therapeutic approach. Nutr Neurosci. 2018;1-12. [Epub ahead of print] [ Links ]
20. Krishnamoorthy U, Dickson M. Maternal phenylketonuria in pregnancy. Obstet Gynaecol. 2005;7(1):28-33. [ Links ]
21. Lee PJ, Ridout D, Walter JH, Cockburn F. Maternal phenylketonuria: report from the United Kingdom Registry 1978-97. Arch Dis Child. 2005;90(2):143-6. [ Links ]
22. Koch R, Hanley W, Levy H, Matalon K, Matalon R, Rouse B, et al. The Maternal Phenylketonuria International Study: 1984-2002. Pediatrics. 2003;112(6 Pt 2):1523-9. [ Links ]
23. Matalon KM, Acosta PB, Azen C. Role of nutrition in pregnancy with phenylketonuria and birth defects. Pediatrics. 2003;112(6 Pt 2):1534-6. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Mariana Abreu
E-mail: marianacgabreu@gmail.com
Agradecimentos
Os autores agradecem à Dra. Elisa Leão, pelo seu contributo no estudo deste caso clínico.
Conflito de interesses
Os autores declaram não ter quaisquer conflitos de interesse.
Fonte de financiamento
Não se registaram quaisquer financiamentos na redação deste artigo.
Recebido em 18-01-2019. Aceite para publicação em 07-07-2019

