











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Aplasia cutis congenita (ACC) is a rare congenital condition characterised by the absence of skin, with or without the absence of underlying structures at the time of birth.1 It mostly affects the scalp (80% - 90%)1 and usually the vertex along embryonic fusion lines, although it can also affect any other body region, such as the face, the torso, and the limbs.2,3
ACC has an estimated incidence of 1 to 3 cases per 10 000 births (although real incidence is hard to ascertain, as it appears to be under-reported since mild cases may go undetected) and is more common in females.1,2,4 The cause is unknown and seems to be multifactorial, with several theories suggesting various risk factors such as: chromosomal abnormalities, trauma, amniotic irregularities, intrauterine complications (vascular events or infection), thrombosis or vascular lesions, and teratogens (misoprostol, benzodiazepines, valproic acid, cocaine, methotrexate, angiotensin-converting enzyme inhibitors, methimazole - the strongest risk factor reported in the literature).1-3 The most widely accepted physiopathological hypothesis states there is a maximum tensile force during rapid brain growth at the ACC region at 10-15 weeks of gestation, which results in a disruption of the overlying skin.3
Frieden Classification, which was proposed in 1986 and continues to be acknowledged by the scientific and medical community, divides ACC into nine groups according to the location and pattern of the skin defect, associated abnormalities, and mode of inheritance (Table 1). This classification highlights the importance of obtaining complete and detailed clinical history of patients with ACC and their families. This enables the clarification of its aetiology and the exclusion of other congenital malformations involving the cardiovascular, gastrointestinal, genitourinary, and central nervous systems.5
Table 1: Frieden Classification of ACC groups.
| Groups | Characteristics | Transmission |
| 1 | ACC located on the scalp without multiple anomalies | Autossomal dominant (AD) or sporadic |
| 2 | ACC located on the scalp with associated limb abnormalities | AD |
| 3 | ACC located on the scalp with associated epidermal and organoid nevi | Sporadic |
| 4 | ACC overlying embryologic malformations | Variable |
| 5 | ACC with associated foetus papyraceus or placental infarcts | Sporadic |
| 6 | ACC associated with epidermolysis bullosa (EB): blistering, usually localised, without multiple congenital anomalies | Depends on the type of EB: AD or autossomal recessive (AR) |
| 7 | ACC localised to extremities without blistering | AD or AR |
| 8 | ACC caused by specific teratogens | Non inherited |
| 9 | ACC associated with malformation syndromes | Variable |
ACC: Aplasia cutis congenita; AD: autossomal dominant; AR: autossomal recessive; EB: epidermolysis bullosa
Case Report
We report a case of a full-term male newborn, from a supervised pregnancy whose mother was treated for hypothyroidism with levothyroxine, without any other complications. No abnormal family history was reported. Prenatal ultrasounds were normal. The infant was born by spontaneous vaginal delivery, at 39 weeks, showing a tight nuchal cord, with no need for resuscitation. Upon examination at the delivery room, aplasia cutis of up to 2 cm of diameter was identified in the occipital region. In the nursery, after a more detailed examination, two adjacent ulcerated lesions with well-demarcated limits were identified in the upper occipital region (20 and 4-5 mm diameter (Fig. 1), with no apparent contiguity on deeper structures). The newborn also presented a 20 mm melanocytic nevus at the dorsal region, toxic erythema, hip click, and a doubtful red reflex eye test. Dermatology observation was requested and a daily Mepilex® absorbent foam dressing was recommended. A transfontanellar ultrasound, that showed no alterations, was suggested to exclude contiguity with central nervous system. Observation by Ophthalmology was normal. The remaining hospitalisation was uneventful and the patient was discharged in the third day of life with partial re-epithelialization of the lesion (Fig. 2), with referrals to Dermatology, Orthopaedics, Genetics, and Neurosurgery Consultations.
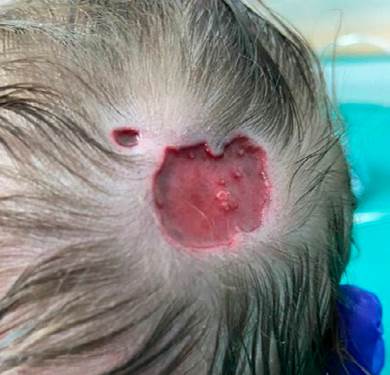
Figure 1: Two adjacent ulcerated lesions with well-demarcated limits in the upper occipital region with no apparent contiguity on deeper structures.
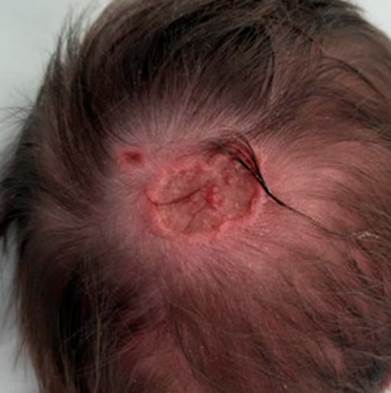
Figure 2: On the third day of life there was a re-epithelialization of the lesion with conservative treatment only.
The patient was observed in a Dermatology Consultation after two weeks. The lesion on the scalp was completely epithelialized, revealing two depressed erythematous plaques with telangiectatic vessels. The family was advised to apply emollient if desquamation occurred. A hairy nevus with 20 mm diameter was described on the right dorsal region. Re-evaluation at the first and sixth months of life revealed complete healing and a new consultation was scheduled for the time when the infant was 18 months old. The patient was seen at the Neurosurgery Consultation, having a cranioencephalic magnetic resonance imaging (MRI-CE) with no alterations. The infant was thus discharged from the consultation and referred to Plastic Surgery Consultation. Hip dysplasia was excluded in the Orthopaedic Consultation. In the Genetic Consultation, minor dysmorphisms were identified: high hair insertion, epicanthus, hypertelorism, rarefaction of the medial third of the eyebrows, depressed nasal bridge, and micrognathia (similarly to the patient’s sister). A normal CGH-array was obtained, and a re-evaluation was scheduled for when the patient turns 18 months of age. In the Plastic Surgery Consultation, excision was scheduled for that same age.
Discussion
In the majority of cases (70%), ACC manifests itself as a solitary defect of the scalp,3 but it occasionally presents as multiple lesions, such as in our case. The lesions are non-inflammatory; well-delimited; may be circular, oval, linear or stellate; membranous or non-membranous; and usually range from 0.5 to 10 cm diameter, but may be larger.1,6 Bone involvement should be excluded, particularly if the skin defect is more than 10 cm diameter. Also the presence of associated malformations and complications (sagittal sinus haemorrhage or thrombosis, local infection or meningitis), which are an important but rare cause of prognosis worsening and death, should be considered.6 There is no consensus on the approach to treat ACC. Since there is great variability in the clinical presentation and severity of ACC, treatment should be individualised according to the size, extent of the defect, presence of associated anomalies, risk of complications, and clinical experience. Treatment options vary from no intervention, conservative treatment or surgery to a combination of surgical and non-surgical approaches.3,7 The conservative treatment consists of regular wound cleansing and application of dressings, topical antibacterial agents, continuous saline drips, along with the use of systemic antibiotics if necessary. The surgical approach includes various procedures, such as primary wound closure, skin grafting, reconstruction of scalp with flaps, and cranial vault reconstruction using bone grafts.1,3
Most small scalp lesions (even if there is underlying bone defect) do not need specific treatment and close spontaneously after weeks or months. Larger defects that may involve underlying dura, with exposure of sagittal sinus, large veins, or brain, may require surgery, either immediately or after conservative treatment, due to the risk of complications.3,6 Although there is agreement that the conservative treatment is the most suitable for smaller lesions, good results have also been achieved when applying this approach to larger lesions. The conservative treatment techniques have increasingly been developed and can currently make use of autologous cultured fibroblasts and keratinocytes or the application of fibroblast growth factors (which accelerate wound healing). This could represent a major change on the approach to ACC, as it reduces the need for immediate surgical interventions.3
In our case, other alterations that could be associated with aplasia cutis were identified at examination and the patient was referred to several consultations. After multidisciplinary assessment, bone and other system involvement were excluded and conservative treatment was kept, with excision scheduled at 18 months old.
In conclusion, after identification of ACC, a complete examination should be performed to exclude other associated congenital malformations. Particular attention should be paid if large skin defects or bone involvement is detected, since these are associated with a higher risk of complications and mortality.

