











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkA previously healthy 23-year-old woman was observed with erythematous and infiltrated plaques associated with significant skin flaccidity forming a pendulous skin fold in the right armpit and a brown macular pigmentation with mild skin flaccidity in the left armpit (Fig. 1). Lesions were asymptomatic and had a 2-year evolution.
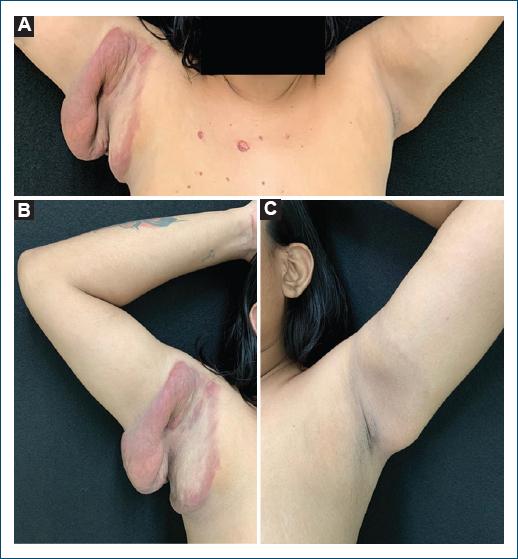
Figure 1 A: skin lesions in both armpits with a 2-year evolution. B: erythematous and infiltrated plaques associated with significant skin flaccidity in the right armpit. C: brown macular pigmentation with mild skin flaccidity in the left armpit.
A skin biopsy of the right armpit revealed a dense dermal granulomatous infiltrate of atypical lymphocytes, neutrophils, histiocytes, plasma cells, eosinophils, and scarce multinucleated giant cells (Figs. 2A-C). Verhoeff staining demonstrated a marked reduction of elastic fibers (Fig. 2D). Immunohistochemistry of the dermal infiltrate showed positivity for cluster of differentiation (CD) 3 and 4 (Fig. 3) and loss of CD7 and CD8 expression, findings that are compatible with the diagnosis of granulomatous slack skin (GSS).

Figure 2 Hematoxylin-eosin-stained sections at ×40, ×200 and ×400, respectively. A: dense granulomatous infiltrate affecting the entire dermis and subcutaneous tissue. B: atypical lymphocytes, neutrophils, histiocytes, plasma cells, eosinophils composing the granuloma. C: scarce multinucleated giant cells. D: verhoeff-stained section at ×600; reduction of elastic fibers.

GSS is a rare variant of mycosis fungoides that mainly affect caucasian men between the third and fifth decades of life1,2.
Histology shows, in addition to elastophagocytosis and emperipolesis, a granulomatous infiltrate mainly composed by atypical lymphocytes, macrophages, and multinucleated giant cells with 20-30 nuclei1-3. This last aspect was not found in our patient and to the best of our knowledge, only another similar case has been reported4. Regarding immunohistochemistry, it demonstrates a cell with a T-helper immunophenotype1, as in the present case, with monoclonal rearrangement of T-cell receptor genes in most tested patients1,2. In addition, some subpopulations of macrophages secrete metalloproteinases that are considered responsible for the degradation and remodeling of the dermal tissue3.
Furthermore, other lymphoproliferative disorders may be present in up to 50% of cases, so patients must be screened and followed up1,2. Due to its rarity, there is no standard treatment, and a complete remission of the disease has seldom been reported4-6.

