











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCTION
Minimal change disease (MCD) is a disorder characterized by podocyte foot process effacement and loss of the glomerular anionic charge. Such lesions cause selective protein leaks (mainly albumin) to the urine, causing a nephrotic syndrome. The disease gets its name from the fact that the damage cannot be seen under a regular microscope, but only using an electron microscope. Primary MCD is the most common cause of nephrotic syndrome in children and young adults.
Although mostly idiopathic, some cases are secondary to infections, drugs, allergies or paraneoplastic syndromes (mostly lymphoproliferative disorders) and therefore it is clinically important to be on the lookout for these conditions. Patients usually present with clinical and laboratorial signs of nephrotic syndrome and normal kidney function, reflecting the subtle changes in nephron structure. The treatment consists in corticotherapy and, for steroid‑resistant patients, calcineurin inhibitors, antimetabolites, cyclophosphamide and even rituximab may be used, depending on the subsequent response to the treatment.
CLINICAL CASE
We present a 21‑year‑old female, sent to the emergency room by her Primary Care Physician in December 2018 for nephrotic range proteinuria on urinalysis (protein 4+, >5g/24h) and generalized oedema associated with nausea and vomiting. The patient denied genitourinary (dysuria, macroscopic haematuria, foamy urine, urine output changes) or systemic symptoms (fever, anorexia, asthenia, weight loss). She had a recent history of non‑steroidal anti‑inflammatory drugs (NSAID) consumption (clonixin 300mg thrice a day for 2 weeks), in the contexto of a worsened chronic hand pain after a car accident. Clinically, the patient was stable and presented with peripheral oedema, without any other relevant findings on her examination. The initial diagnostic workup showed normal kidney function (serum creatinine (SCr) 0.65mg/dL), hypoalbuminaemia (Albumin 1.6g/dL), hypercholesterolaemia (Total cholesterol 379mg/dL), negative C Reactive Protein (CRP) and proteinuria on both urine dipstick (protein +++) and urinalysis (30g/dL). A kidney Doppler ultrasound was performed, showing a diffuse increase in cortical reflectivity, loss of parenchymal differentiation and no signs of renal vein thrombosis. We conducted a full auto‑immune, immunological and serological study and admitted the patient to the Nephrology ward on intravenous diuretics. During the following days, the patient maintained a good urine output, with partial resolution of the oedema. By the third day on the ward, we performed a percutaneous kidney biopsy to clarify the pathological process. Soon after, the blood and urine tests came up and are shown on Table 1.
Table 1 Initial diagnostic workup
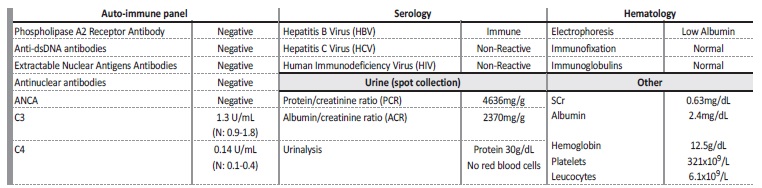
Anti-dsDNA antibodies: anti double stranded DNA antibodies; ANCA: anti-neutrophil cytoplasmic autoantibody; SCr: Serum creatinine
The diagnostic workup was inconclusive: no secondary (such as auto‑immune diseases and chronic viral infections) or primary (Primary Membranous Nephropathy) causes for the nephrotic syndrome were identified. Serum electrophoresis and immunofixation did not reveal any monoclonal component and, as such, a dysproteinaemia was virtually ruled out. The spot urine collection showed benign urinary sediment and confirmed the nephrotic range proteinuria, mainly constituted by albumin. Suspecting Minimal Change Disease (MCD), we started prednisolone (PDN) 1mg/kg/day, with good clinical response and attenuation of urinary protein losses. The biopsy results were soon available: on optical microscopy, the pathologist identified seven glomeruli with normal morphology (without mesangial/capillary hypercellularity or cellular crescents) (Figure 1‑ A and B), small caliber arteries with minimal intimal fibrosis and small areas of interstitial fibrosis; immunofluorescence was performed on frozen tissue and no imune deposits were found. The biopsy results were compatible with a MCD diagnosis and so, the patient was discharged with PDN 60mg and advised to follow a hydric restrictive (500‑1000mL/day)/low‑sodium diet. We also prescribed apixaban 2.5mg twice a day and losartan 50mg once a day to prevent venous thromboembolism and reduce proteinuria, respectively.
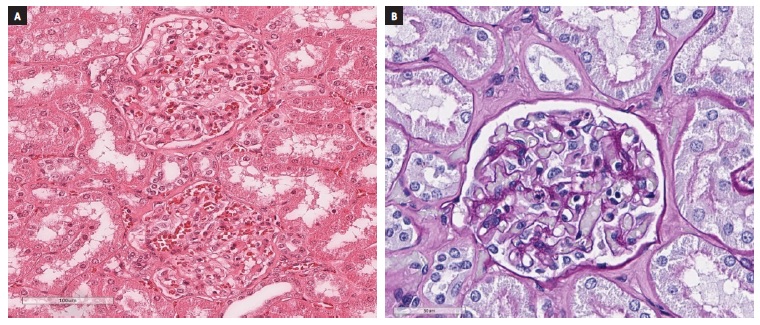
Figure 1 Kidney biopsy fragment on optical microscopy, using haematoxylin-eosin (A) and Periodic acid-reactive Schiff (B) colorations. The glomeruli show normal morphology, without mesangial/capillary hypercellularity or cellular crescentes
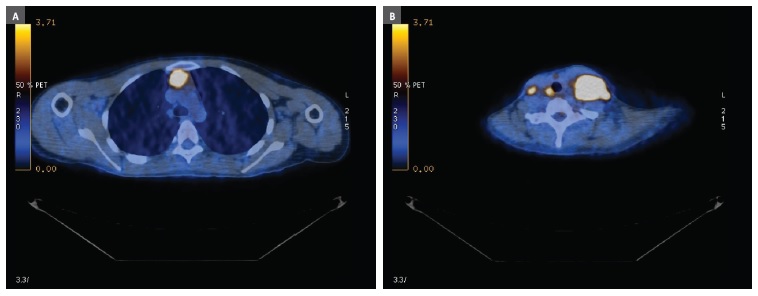
Figure 2 PET results: high-grade metabolic lymphoid mediastinal mass (A) and bilateral cervical adenopathies (B)
By the time of her Nephrology appointment, there were only traces of proteinuria on urinalysis (10g/dL) and, as expected, her Protein/Creatinine Ratio (PCR) had already improved (0.06mg/g). The patient remained on PDN 60mg for 15 days, and then was started on a corticoid taper. On the following months there were no signs of a relapse, as her kidney function and PCR remained under control, eventually stopping PDN after six months.
Two months later, the patient noticed a left cervical adenopathy, which grew progressively and extended to other sites: first to the right cervical and then to the inguinal region. Besides local discomfort, the patient also complained of nocturnal hyperhidrosis, but denied weight loss, anorexia, asthenia or other B symptoms. On physical examination the patient presented with a left cervical mass (diameter > 5cm) and multiple adenopathies on the cervical and inguinal region, with fibroelastic consistency. Her blood work showed no signs of systemic infections (HIV, HBV, HCV, Syphilis and parasites) and the haemogram and serum electrophoresis were normal. The Haematology consultant requested an ultrasound‑guided mass biopsy, showing a lymphoid cell proliferation, positive for CD30, CD15, MUM‑1 and BCl6 and negative for CD45, CD3 and CD79‑alpha.
The biopsy results were compatible with a classic Hodgkin’s Lymphoma diagnosis (nodular sclerosing, grade I). A Positron emission tomography (PET) was performed, revealing a high‑grade metabolic lymphoid mediastinal mass (45mm diameter; standardized uptake value 8.7) and supra‑diaphragmatic lymph node evolvement (Figure 2-A and B).
The treatment consisted in a combination of radiotherapy (30 Gy) and four cycles of chemotherapy across 6 months (ABVD‑doxorubicin, bleomycin, vinblastin, dacarbazine). In the following months her clinical status improved and the cervical mass had a significant size reduction.
As for MCD, the disease remained in remission, without therapeutic adjustments. A post‑treatment PET scan was requested, showing a residual mediastinal mass with a class 3 Deauville (uptake > mediastinum but < liver).
DISCUSSION
It is postulated that MCD is a disorder of T cells, which release a cytokine that injures the glomerular epithelial foot processes, leading to a decreased synthesis of polyanions. The polyanions constitute the charged barrier to the filtration of macromolecules, such as albumin. When the polyanions are damaged, leakage of albumin follows. The identity of this circulating permeability factor is uncertain, although it is postulated that haemopexin may play a role in podocyte damage.1 Many cytokines and proteins, like Interleukin (IL) 4, IL 12, IL13, CD80 and nephrin, have been associated to MCD and can also contribute to the damage. In a recent study, published in the Journal of the American Society of Nephrology, Watts et al. state they have identified circulating nephrin autoantibodies during active disease that were significantly reduced or absent during treatment response in a subset of patients with MCD, suggesting the need for a new molecular classification of MCD.2
MCD is responsible for 70‑90% and 10% of nephrotic syndrome cases in children/young adults and elder adults, respectively. In children, males are twice more affected than females, with equal gender distribution in adults. MCD is usually idiopathic, especially in children.3 Secondary forms may be due to viral infections (Syphilis, HIV, Hepatitis), drugs (NSAIDs, lithium, some antibiotics, bisphosphonates), allergic reactions, immunologic diseases or neoplasia4 The diagnosis is suggested by clinical and laboratorial features of nephrotic syndrome: hypoalbuminaemia, nephrotic range proteinuria, hypercholesterolaemia and peripheral/peri‑orbitary oedema.
At presentation, 50‑75% of the patients have a normal kidney function, reflecting the subtle changes in nephron structure. Early markers of the disease are under investigation: CD80, a transmembrane protein, is present on podocytes and urinary CD80 levels appear to be significantly elevated in MCD, but not in FSGS or other glomerulopathies.4,5 However, further studies need to be conducted to assess the efficacy of these new biomarkers. In children a suspicion diagnosis can be made solely on clinical and laboratorial findings and treatment can be started empirically. In non‑responsive children and in adults, like our patient, the differential diagnosis is made with entities cursing with nephrotic syndrome such as focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN) (primary or secondary) and diabetic nephropathy. This case depicted a young female with classic nephrotic syndrome findings: generalized oedema, nephrotic range proteinuria, hypoalbuminaemia and hypercholesterolaemia. The diagnostic workup did not reveal any primary or secondary causes (such as auto‑immune diseases or chronic viral infections) for the nephrotic syndrome. A negative anti‑PLA2R antibody, although it does not exclude, suggested other entity rather than MN, because is usually positive in 95% of the patients. Furthermore, the findings on the kidney biopsy were compatible with a MCD diagnosis and did not suggest another pathological process. The patient denied constitutional symptoms like fever, nocturnal hyperhidrosis, weight loss or any focalizing complaints and, for that reason, the hypothesis of a neoplastic process was set aside and a full body CT scan was not performed.
In addition, the patient had a history of recent heavy NSAID consumption, prior to developing symptoms, and therefore, despite the absence of acute interstitial nephritis that would give further support to this hypothesis, this case was interpreted as a MCD secondary to NSAIDs.
The treatment of MCD, according to Kidney Disease Improving Global Outcome (KDIGO) guidelines, consists of corticoid therapy, usually PDN 1mg/kg/day until 1 week after complete remission is achieved, with a minimum duration of 4 weeks and a maximum of 12‑16 weeks. It is the recommended therapy even in the context of an identified likely trigger, such as NSAID consumption. Complete remission is defined by KDIGO as a urinary PCR < 30 mg/g or a urine dipstick negative for protein and is obtained in 85‑90% of the affected children and in approximately 80% of the adults. [4,6] This treatment is followed by a corticoid taper over a few months. In our case, the patient achieved complete remission in 6 weeks with PDN 60mg/day, followed by progressive dose reduction over 6 months, with no relapses in the meantime.
However, soon after stopping PDN the patient developed a bilateral cervical mass, associated with nocturnal hyperhidrosis. A mass biopsy was performed and the results were compatible with a Hodgkin’s Lymphoma (HL) diagnosis. The patient was started on radio and chemotherapy for 6 months with significant clinical and imagiological improvement. Although it is well recognized that many types of glomerulonephritis are associated with classical Hodgkin’s disease, the incidence of nephrotic syndrome (NS) is low and estimated at about 0.5 to 1%.7,8 In two studies of Hodgkin’s disease patients, two glomerulopathies were significantly associated with HL: amyloidosis (0.1% of cases) and MCD (0.4% of cases).8 The pathogenesis of this association remains poorly understood and the underlying molecular link is still unknown, although alteration in T‑lymphocyte functions might be the causal factor.8 There is a close relationship between the courses of HL and MCNS, particularly in that remission of MCNS occurs after successful treatment of HL, suggesting that MCD is a paraneoplastic syndrome in the contexto of HL. However, current evidence shows that MCD may precede, occur simultaneously with, or follow a broad spectrum of non‑Hodgkin lymphoproliferative disorders, mainly with neoplasms originating from B cells (94.4% of cases).9 In our case, one can speculate whether HL was already present by the time the diagnosis of MCD was made, that was kept under control and asymptomatic due to a long course of corticotherapy or an occasional finding of a MCD patient that eventually developed a HL (the absence of recurrence of the nephrotic syndrome during the six month period that preceded the HL diagnosis would support this last hypothesis). In either case, it is important to keep this association in mind when presented with a MCD case, enquire for B‑symptoms and carefully inspect for adenopathic masses in every consultation.
KEY MESSAGES
1. Minimal change disease (MCD) is the most common cause of nephrotic syndrome in children (70‑90%) and is responsible for 10% of the cases in adults.
2. Although usually idiopathic, it can also be secondary to viral infections, drugs (NSAIDs, lithium, some antibiotics, bisphosphonates) allergic reactions, immunologic diseases or neoplasia (Leukaemia, Hodgkin Lymphoma).
3. In adults the differential diagnosis is made with entities cursing with nephrotic syndrome such as FSGS and MN (primary or secondary)
4. The treatment is based on high dose corticotherapy for 4‑16 weeks until complete remission is achieved. In frequent relapsers or corticoid resistant patients’ other immunosuppressive therapies can be added.
5. MCD may precede, occur simultaneously with, or follow a broad spectrum of non‑Hodgkin lymphoproliferative disorders, mainly with neoplasms originating from B cells

