











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1. Introducción
El análisis de secuencias biológicas es una de las actividades fundamentales en bioinformática (Dimitrios Vasileiou, 2023) facilita la genómica estructural, comparativa y funcional, sirviendo como marco subyacente para predicciones sistemáticas de estructura/función, clasificación y anotación de proteínas. Este análisis ayuda a identificar funciones o características cruciales de los organismos (Katie Emelianova, 2023) (Azza E. Ahmed, 2021), como la complejidad de la regulación transcripcional y la información molecular, entre otras características o información valiosa.
Ante la gran cantidad de datos biológicos, es imperativo recurrir a software eficiente y probado que facilite estos análisis, más aún cuando se ejecutan a gran escala porque algunas de las herramientas actuales son complejas de utilizar (Azza E. Ahmed, 2021).
En este contexto, se ha identificado que existe una amplia variedad de software bioinformático como BioPython (Cock, 2009), cogent3 (Cogent3 Team), cactus (Joel Armstrong, 2020), PyCogent (Rob Knight, 2007), PyEvolve (Andrew Butterfield, 2004), entre otros; que realizan tareas específicas, y que a su vez promueven la dispersión de herramientas. Como resultado, profesionales relacionados al campo encuentran ineficiencias en sus tareas diarias al utilizar varios softwares en lugar de uno.
Jean-Baptiste Cazier en 2013 realizó una encuesta en donde obtuvo 47 respuestas de expertos en el campo (Cazier, 2013), en donde plantea la misma problemática sobre la existencia de muchas herramientas bioinformáticas, como resultado identificó los siguientes puntos:
89% de las respuestas coinciden en que hay demasiadas herramientas bioinformáticas.
34% de las opiniones coinciden en que es difícil encontrar software adecuado para tareas específicas.
30% mencionan que es necesario compartir experiencias y conocimientos para hacer un mejor uso de las herramientas disponibles.
13% de las opiniones indicaron que es necesario hacer un esfuerzo conjunto por unificar las herramientas en un solo entorno.
Otro aspecto relevante que se obtuvo de esta encuesta es que los expertos encuentran muchas dificultades en el uso de las herramientas bioinformáticas existentes, esto se presenta en los siguientes puntos:
25% de las opiniones mencionan dificultad para encontrar software adecuado para tareas específicas.
13% mencionan la falta de un esfuerzo conjunto para unificar las herramientas en un solo entorno.
15% indicaron la complejidad y la curva de aprendizaje asociadas con algunas herramientas ante la necesidad de tener conocimientos de programación o de compilación de archivos fuente.
11% resaltaron los problemas de documentación y falta de comprensión de los algoritmos subyacentes en algunas herramientas.
15% mencionan la proliferación de herramientas que genera la confusión sobre cuál utilizar.
17% indican la necesidad de actualización y mantenimiento continuo de las herramientas.
9% mencionan la falta de recursos para evaluar y comparar las herramientas disponibles.
15% indicaron las dificultades en la instalación y configuración de algunas herramientas.
15% de los expertos coinciden en que existe variabilidad en los resultados obtenidos con diferentes herramientas para la misma tarea.
Ante este contexto este articulo presenta una propuesta de aplicación web con el objetivo de unificar diferentes herramientas de análisis en un mismo entorno enfocado en el análisis elemental y sin código que conjunte en un solo entorno diferentes herramientas atendiendo las problemáticas identificadas.
2. Plataformas relacionadas
Para identificar las funcionalidades que se pueden implementar dentro de la propuesta de unificación de herramientas se realizó una investigación explorando las principales plataformas científicas relacionadas al campo que incluyen diferentes herramientas, algunas de las principales plataformas identificadas son:
En la siguiente subsección se presentan las funciones que cubre cada una de estas plataformas.
2.1. Expasy
Ofrece una amplia variedad de funciones relacionadas con el análisis y la manipulación de datos biológicos, dentro de las cuales están:
Análisis de Secuencias: Capacidad para analizar secuencias biológicas.
Predicción de Estructuras: Previsión de la estructura tridimensional de proteínas y ácidos nucleicos.
Búsqueda de Homologías: Identificación de secuencias similares en bases de datos.
Diseño de Primers: Herramientas para el diseño eficiente de cebadores para PCR.
Análisis de Expresión Genética: Evaluación de la expresión génica a nivel transcriptómico.
Anotación Funcional: Asignación de funciones biológicas a secuencias.
Bases de Datos: Acceso a una variedad de bases de datos biológicas.
2.2. NCBI Tools
La suite de herramientas de NCBI ofrece una amplia gama de recursos para la investigación biomédica:
Bases de Datos Biomédicas: Repositorios de datos biomédicos para investigación y análisis.
BLAST: Herramienta para comparar secuencias biológicas con bases de datos.
Entrez: Sistema de búsqueda y recuperación de información biomédica.
Información de Genes: Recursos para acceder y comprender información genética.
Exploración de Genomas y Especies: Herramientas para explorar información genómica.
2.3. Chimera
Chimera se centra en la visualización y modelado de estructuras moleculares:
Visualización de Estructuras Moleculares: Representación visual de estructuras tridimensionales.
Modelado de Estructuras: Creación y manipulación de modelos tridimensionales de macromoléculas.
Análisis de Interacciones: Evaluación de interacciones moleculares.
Renderización de Imágenes: Generación de imágenes de alta calidad para la presentación de datos.
Análisis de Archivos PDB: Exploración y análisis de datos estructurales en formato PDB.
2.4. BLAST
BLAST es una herramienta ampliamente utilizada para comparar secuencias biológicas:
Comparación de Secuencias: Identificación de similitudes entre secuencias biológicas.
Búsqueda Rápida en Bases de Datos: Acceso rápido a información relevante en bases de datos.
Alineación de Secuencias: Alineación de secuencias para identificar regiones conservadas.
Variantes y Mutaciones: Análisis de variaciones genéticas y mutacionales.
BLASTp, BLASTn, etc.: Variantes de BLAST para diferentes tipos de secuencias.
Considerando las funcionalidades identificadas en cada plataforma y con el objetivo de unificar la mayor cantidad de funcionalidades posibles, se realizó una comparativa entre ellas y la funcionalidad que cubren (ver Tabla 1).
Tabla 1 Comparativa respecto a la funcionalidad cubierta de cada plataforma.
| Funcionalidad | Expasy | NCBI Tools | Chimera | BLAST |
| Análisis de secuencias | ✔️ | ✔️ | ||
| Predicción de estructuras | ✔️ | ✔️ | ||
| Búsqueda de homologías | ✔️ | |||
| Diseño de primers | ✔️ | |||
| Análisis de expresión genética | ✔️ | |||
| Anotación funcional | ✔️ | |||
| Bases de datos | ✔️ | ✔️ | ||
| Cálculo y generación de árboles filogenéticos | ✔️ | |||
| Análisis de archivos PDB y visor de PDB | ✔️ | |||
| Traducción y transcripción de secuencias | ||||
| BLAST (BLAST-P, BLAST-N, BLAST-X, TBLASTN, TBLASTX) | ✔️ | ✔️ | ||
| Entrez | ✔️ | |||
| Información de genes | ✔️ | |||
| Archivos de artículos científicos | ✔️ | |||
| Exploración de genomas y especies | ✔️ | |||
| Transcripción inversa | ✔️ | |||
| Análisis de Propiedades de Proteomas y ADN | ✔️ | |||
| Alineación y traducción de secuencias | ✔️ | |||
| Análisis de interacciones | ✔️ | |||
| Renderización de imágenes | ✔️ | |||
| Variantes y mutaciones | ✔️ |
Con base a la anterior comparativa, se identifica que del total de funcionalidades identificadas la plataforma que cubre mayor cantidad de esas funciones es NCBI Tools con un 43% de cobertura. De acuerdo al resultado de la Tabla 1, se establecieron las funcionalidades que debería tener la herramienta web que permitiría unificar diversas funcionalidades estableciendo que la plataforma propuesta cubrirá con 52% del total de las funcionalidades, tal como se ve en la Tabla 2, los signos de guión significan que la propuesta cubre parcialmente con esa funcionalidad:
Tabla 2 Cumplimiento de funcionalidades de la propuesta de plataforma.
| Funcionalidad | Expasy | NCBI Tools | Chimera | BLAST | Propuesta |
| Análisis de secuencias | ✔️ | ✔️ | ✔️ | ||
| Predicción de estructuras | ✔️ | ✔️ | |||
| Búsqueda de homologías | ✔️ | ||||
| Diseño de primers | ✔️ | ➖ | |||
| Análisis de expresión genética | ✔️ | ✔️ | |||
| Anotación funcional | ✔️ | ||||
| Bases de datos | ✔️ | ✔️ | ✔️ | ||
| Cálculo y generación de árboles filogenéticos | ✔️ | ✔️ | |||
| Análisis de archivos PDB y visor de PDB | ✔️ | ✔️ | |||
| Traducción y transcripción de secuencias | ✔️ | ||||
| BLAST | ✔️ | ✔️ | ✔️ | ||
| Entrez | ✔️ | ➖ | |||
| Información de genes | ✔️ | ✔️ | |||
| Archivos de artículos científicos | ✔️ | ||||
| Exploración de genomas y especies | ✔️ | ||||
| Transcripción inversa | ✔️ | ✔️ | |||
| Análisis de Propiedades de Proteomas y ADN | ✔️ | ✔️ | |||
| Alineación y traducción de secuencias | ✔️ | ✔️ | |||
| Análisis de interacciones | ✔️ | ||||
| Renderización de imágenes | ✔️ | ||||
| Variantes y mutaciones | ✔️ |
La siguiente sección aborda la propuesta de unificación, así como la selección de tecnologías que ayudan a lograr unificar las funcionalidades en un solo entorno sin necesidad de que el usuario tenga conocimientos de programación o de compilación de archivos.
3. Propuesta de unificación
Se consideró un conjunto completo de software y herramientas bioinformáticas ya existentes que cuentan con una amplia gama de herramientas que facilitan el análisis de secuencias biológicas, como ExPASy (Gasteiger, 2003) y NCBI Services (Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information, 2017), con el objetivo de identificar las funciones que se pudieran unificar en un mismo entorno.
Esta propuesta de herramienta fue desarrollada utilizando Python como lenguaje de programación, misma que integra librerías que han sido probadas como: BioPython (Cock, 2009), BioPandas (Raschka, 2017) y 3Dmol (Rego, 2014). Mismas que fueron seleccionadas por su estabilidad, confiabilidad y funcionamiento extensivo en el análisis de secuencias biológicas.
3.1. Arquitectura de la aplicación
La arquitectura de la aplicación está estructurada para optimizar la integración de varias funcionalidades de diferentes librerías de Python bajo el framework DJANGO, por ello esta propuesta reúne herramientas que se enfocan en los siguientes puntos:
Análisis de secuencias de ADN (ver Figura 1, con la secuencia de entrada “ACGTAAACGT”).
Análisis de propiedades: recibe como entrada una secuencia de nucleótidos, como resultado entrega: secuencia complementaria, secuencia complementaria inversa, distribución de los aminoácidos presentes con su gráfico.
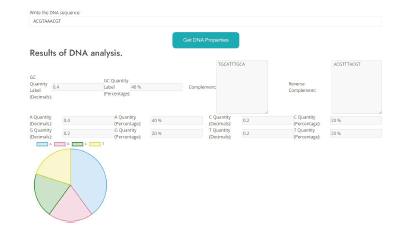
Como resultado nos entrega una cantidad de GC de 0.4, equivalente al 40% porcentualmente.
Adicionalmente nos entrega su secuencia complementaria “TGCATTTGCA” y su secuencia reversa complementaria “ACGTTTACGT”.
Finalmente nos entrega la distribución de los elementos de la secuencia en decimales y porcentualmente A 40%,GCT con una distribución del 20% cada uno.
Análisis de secuencias proteómicas (ver Figura 2, con la secuencia de entrada MEQPATKRRKQHNENLETEPTGADYERMSAWLQREGADFAN VTIHKSHESEGYGIYAARA).
Análisis de propiedades: recibe como entrada una secuencia de proteínas y un pH, como resultado entrega: tamaño de la secuencia, peso molecular, aromaticidad, índice de inestabilidad, punto isoeléctrico, estructura secundaria, coeficiente de extinción molar, numero de puentes de disulfuro, índice de hidropaticidad, carga eléctrica e hidrofobicidad.

Como resultados entrega la longitud de la secuencia: 60, peso molecular: 6878.4238 g/mol, aromaticidad: 0.083, índice de inestabilidad: 64.68, entre otros.
Transcripción y transcripción inversa (ver Figura 3, con la secuencia de entrada de GTGAAAAAGATGCAATCTATCGTACTCGCACTTTCCCTGGTTC TGGTCGCTCCCATGGCAGCACAGGCTGCGGAAATTACGTTAGTCCCGTC AGTAAAATTACAGATAGGCGATCGTGATAATCGTGGCTATTACTGGGATGGAGGTCACTGGCGCGACCACGGCTGGTGGAAACAACATTATGAATGGCGAGGCAATCGCTGGCACCTACACGGACCGCCGCCACCGCCGCGCCACCATAAGAAAGCTCCTCATGATCATCACGGCGGTCATGGTCCAGGCAAACAT CACCGCTAA).
Transcripción: recibe como entrada una secuencia de nucleótidos, como salidas entrega la transcripción de la secuencia a mRNA y hebras plantilla.
Transcripción inversa: recibe como entrada una secuencia de mRNA y como salida entrega la secuencia de nucleótidos.
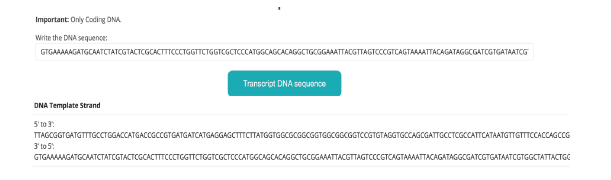
En la figura se aprecia el resultado, nos entrega la cadena completa de 5’ a 3’ y de 3’ a 5’.
Traducción de secuencias (ver Figura 4, con la secuencia de entrada AUUU UCUUUGCUCUUGAGCUCUGGCACUUCUCUGCUGCUGUCUG).
Como entradas recibe secuencias de mRNA, selecciona la tabla de codón a utilizar y como salida entrega el tipo de secuencia y la traducción de mRNA a ADN de la secuencia.
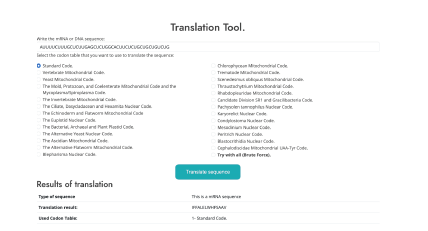
Como resultados nos entrega, el tipo de secuencia de entrada, el resultado de la traducción de la secuencia que es: “IFFALELWHFSAAV” y la tabla de codones utilizada para la traducción: tabla de codón estándar.
Alineación de secuencias por pares (ver Figura 5, con secuencia de entrada 1: “ATTTTCTTTGCTCTTGAGCTCTGGCACTTCTCTGCTGCTGTCTG” y secuencia de entrada 2: “ATTTTCTTGCTCTTGAGCTCTGGCACTTCTCTG CTGCTGTC”).
El usuario ingresa dos secuencias que quiera alinear, seleccionando también el modo de alineación, si es global o local y como salida la herramienta entrega estadísticas del proceso de alineación, así como las secuencias alineadas.
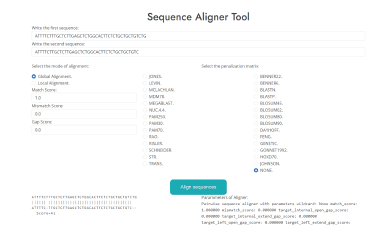
Como resultados nos entrega la secuencia alineada y las estadísticas del alineador (puntaje de coincidencia, puntaje de no coincidencia, tipo de alineamiento, entre otros).
Búsqueda de secuencias.
El usuario proporciona una secuencia que puede ser proteína, nucleótido o gen, la aplicación hace una búsqueda utilizando BLAST y como salida entrega una lista de organismos que comparten similitudes con la secuencia. De esta funcionalidad no se coloca imagen por la extensión de esta.
Visualizador de archivos PDB (ver Figura 6, como valores de entrada recibe un archivo PDB que puede ser descargado en cualquier portal que ofrezca este tipo de visualizaciones de las proteínas de los organismos).
La herramienta recibe como entrada un archivo PDB, y como salida entrega la estructura 3D de la proteína con sus diferentes modos de visualización.

Como resultados entrega la visualización, así mismo cuenta con varios modos de visualización (palos, caricatura, esferas, vectores, etc.) dentro de los cuales el usuario puede interactuar para cambiar el modo de visualización.
Árboles filogenéticos (ver Figura 7).
La herramienta recibe como entrada un archivo en formato CLUSTAL, como salidas entrega el cálculo de distancias entre las secuencias biológicas y genera un árbol filogenético.

Como resultado, entrega el cálculo de distancias entre cada rama de las secuencias del archivo CLUSTAL, un diagrama que permite visualizar las distancias de las secuencias y el texto que forma cada rama del diagrama.
Análisis de MOTIFS y expresiones regulares (ver Figura 8, con las secuencias de entrada: TACAA, TACGC, TACAC, TACCC, AACCC, AATGC, AATGC).
Análisis de MOTIFS: El usuario recibe una lista de MOTIFS, como salidas entrega una matriz de conteo, la secuencia de consenso, secuencia de consenso degenerado, reversa complementaria y la secuencia de consenso complementaria degenerada.
Expresiones regulares: la herramienta recibe una expresión regular de lo que va a buscar globalmente en la base de datos que se le cargue.
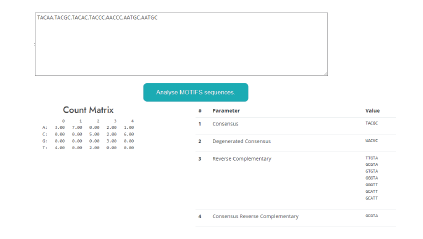
Como resultados nos entrega la matriz de conteo, la secuencia consenso, la secuencia de consenso degenerado, la secuencia reversa complementaria y la secuencia consenso reversa complementaria.
Conexión con base de datos de hongos y algas con datos provenientes del JGI (JGI, 2022) o de cualquier otra fuente.
El sistema se conecta a una base de datos MONGODB que debe ser instalada previamente, misma que contiene todas las secuencias que el usuario decida cargar ya sea de hongos, algas o del tipo de organismo que el usuario trabaje.
En las subsecciones siguientes se aborda la interfaz de usuario, el alcance y adaptabilidad, y otros aspectos relevantes para el desarrollo de esta propuesta de herramienta.
3.2. Interfaz de usuario
Como se mencionó inicialmente, la propuesta de herramienta posibilita a que usuarios sin conocimiento en programación puedan usar este tipo de herramientas, por ello el desarrollo de una interfaz de usuario intuitiva fue necesaria para lograr este cometido (ver Figura 9).

La Figura 9 muestra la interfaz de usuario de la propuesta de herramienta con sus herramientas que están integradas dentro de la propuesta de aplicación.
3.3. Alcance y adaptabilidad
Esta propuesta de herramienta está diseñada para analizar secuencias biológicas de organismos de hongos y algas. Sin embargo, el software se adapta a una gama más amplia de secuencias biológicas sin importar si es o no de este tipo de organismos, lo que destaca su versatilidad. La adaptabilidad de la herramienta garantiza su utilidad en diversos contextos de investigación más allá del alcance inicial.
El tipo de secuencias que pueden analizar con la propuesta son secuencias genómicas y proteómicas de cualquier organismo, las secuencias genómicas son secuencias de nucleótidos o bases en una molécula de ADN (Brown, 2002). Por otra parte, las secuencias proteómicas son el total de moléculas proteicas presentes en una célula, tejido u órgano (Maino, 2000).
El alfabeto de las secuencias genómicas está conformado por 4 elementos: A, C, G y T; mientras que el alfabeto de las secuencias proteómicas se conforma por 26 elementos.
3.4. Optimización del flujo de trabajo
La integración de diferentes componentes de software en un mismo entorno es un punto clave del desarrollo de esta herramienta, ya que esta integración agiliza el flujo de trabajo eliminando la necesidad del usuario de navegar entre diferentes aplicaciones o software diferentes para el uso de sus herramientas o de tener conocimientos de programación.
4. Resultados
La propuesta de herramienta presentó desafíos en la integración de tecnologías y componentes dentro de un mismo entorno principalmente por la diferencia entre los lenguajes de programación. Sin embargo, gracias al uso de diferentes tecnologías se logró una adecuada integración. A continuación, se presentan los resultados obtenidos, destacando cómo el uso de estas librerías ha contribuido a desarrollar la funcionalidad integral del software.
4.1. Integración de funcionalidades
La combinación de diferentes librerías en el desarrollo de la herramienta ha permitido la integración fluida de una amplia gama de funcionalidades relacionadas con el análisis de secuencias biológicas. Desde el análisis de propiedades de secuencias de ADN y proteínas hasta la generación de árboles filogenéticos, cada herramienta se ha implementado de manera coherente dentro del entorno unificado del software.
La herramienta provee de resultados cuantitativos de los resultados que entrega, por ejemplo utilizando el generador de árboles filogenéticos muestra el cálculo de las distancias entre las secuencias alineadas (ver Figura 10 esquina superior izquierda). Otro resultado cuantitativo que presenta la plataforma es al usar el alineador de secuencias por pares, ya que presenta el puntaje de alineamiento y una sección de estadísticas del alineador (ver Figura 10 sección inferior). Otro ejemplo de datos cuantitativos es al usar la herramienta de MOTIFS, ya que nos entrega una matriz de conteo de proteínas (ver Figura 10 esquina superior derecha).

4.2. Eficiencia y Precisión
El uso de librerías especializadas ha garantizado la eficiencia y precisión en el análisis de las secuencias biológicas. Por ejemplo, la implementación de BLAST ha permitido realizar búsquedas rápidas y precisas en la base de datos de NCBI, identificando secuencias similares con alta fiabilidad, funcionalidad que ha sido validada en el artículo publicado por P. Cock (Cock, 2009) que aborda la funcionalidad de BioPython. Asimismo, la integración de algoritmos de alineación de secuencias ha facilitado la comparación y el análisis de similitudes entre secuencias de ADN y proteínas que al igual esta funcionalidad ha sido validada por expertos en el campo que desarrollaron cada componente que se integró a esta herramienta.
4.3. Facilidad de uso
Gracias a la utilización de librerías bien establecidas y documentadas, unido al framework de DJANGO con sus integraciones la herramienta desarrollada ofrece una interfaz amigable y fácil de usar para los usuarios, destacando que se hizo más accesible el uso de herramientas bioinformáticas sin la necesidad de tener conocimientos en programación. Las funciones se presentan de manera clara, accesible y bien documentadas, lo que permite a los usuarios realizar análisis mediante el uso de los componentes de la herramienta de manera intuitiva y eficiente (Ver Figura 9).
4.4. Escalabilidad y adaptabilidad
La arquitectura del sistema y la forma en la que fue desarrollada la aplicación garantiza su escalabilidad y adaptabilidad a futuras actualizaciones y ampliaciones de funcionalidades ya que cada componente es independiente uno del otro, por tanto se pueden desarrollar más módulos mismos que se pueden integrar fácilmente a la herramienta sin comprometer otros componentes o funcionalidades ya desarrolladas, de esa forma se garantiza una constante actualización y adaptabilidad a futuras tecnologías, que es otra de las problemáticas identificadas por los expertos ante la disponibilidad de muchas herramientas que no reciben actualización.
5. Conclusiones
En esta articulo se destaca la diversidad de herramientas bioinformáticas disponibles que presentan tanto ventajas como desafíos significativos respaldados por la opinión de expertos en el campo. Si bien esta variedad ofrece una amplia gama de funcionalidades, también plantea problemas de eficiencia y efectividad para los profesionales, como la dificultad para encontrar software adecuado y la falta de unificación en las herramientas disponibles.
En respuesta a estas problemáticas identificadas, se propone una solución innovadora: el desarrollo de una aplicación web que unifica diversas funcionalidades para análisis de secuencias biológicas. Esta propuesta surge como una respuesta directa a la necesidad de simplificar y optimizar el proceso de análisis, ofreciendo a los usuarios un entorno integrado que abarca múltiples funcionalidades sin requerir conocimientos de programación.
La elección de tecnologías sólidas, como Python y el framework Django, junto con la integración de librerías especializadas como BioPython, BioPandas, 3Dmol.js, BLAST, entre otras, proporciona una base robusta para el desarrollo de la herramienta propuesta. Aunado a la elección de una arquitectura modular, misma que asegura la escalabilidad y adaptabilidad de la aplicación a futuras actualizaciones y ampliaciones de funcionalidades sin comprometer funcionalidades ya implementadas.
La interfaz de usuario intuitiva y la documentación exhaustiva son aspectos clave de la propuesta, ya que facilitan la adopción y el uso adecuado de la herramienta por parte de usuarios de diversos niveles de experiencia. La capacidad de proporcionar resultados cuantitativos, respaldada por la validación de la eficacia de la herramienta, fortalece su utilidad y confiabilidad en el ámbito científico.
Esta propuesta de herramienta representa un avance significativo en la simplificación y mejora de los procesos de análisis de secuencias biológicas en el campo de la bioinformática. Su implementación promete no solo mejorar la eficiencia y efectividad de los profesionales en este campo, sino también impulsar el progreso científico y tecnológico en áreas afines.
6. Trabajo Futuro
A pesar de los avances significativos logrados con nuestra propuesta de herramienta, existen áreas que requieren atención adicional y mejoras para maximizar su utilidad y efectividad en el campo de la bioinformática. En este sentido, se identifican varias líneas de trabajo futuro que podrían enriquecer y fortalecer aún más nuestra herramienta: a) incluir la integración de algoritmos avanzados de análisis de secuencias, herramientas de visualización de datos mejoradas y métodos de análisis predictivo-basados en aprendizaje automático; b) expandir la herramienta para abarcar una gama más amplia de organismos, desde la medicina hasta la agricultura y la conservación de la biodiversidad.

