











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a highly fatal condition that is being increasingly recognized in adults. Diagnosis and management remain challenging. Fever, cytopenias, coagulopathy, and hepatosplenomegaly are hallmark findings. Identifying the trigger event is crucial but can be quite difficult due to the diverse presentations and the scarce clinical experience of most physicians with this syndrome. The primary treatment goal is to eradicate the underlying precipitating factor and suppress the massive inflammatory response with steroids, immunoglobulins, or immunomodulators.
Case Report
A 23-year-old woman with ulcerative colitis diagnosed in 2018, Montreal type E3, presented to the hospital with fever and arthralgias. She had received a first dose of the COVID-19 mRNA vaccination (BNT162b2 mRNA vaccine) 15 days previously. Management of her ulcerative colitis had been complicated in the past by the development of HLH secondary to cytomegalovirus (CMV) infection in 2019, treated with antiviral therapy (ganciclovir/valganciclovir). She had been in remission since then under vedolizumab therapy. She had no other comorbidities and medications besides vitamin D supplementation. She is a social worker and had an unremarkable travel history and unknown family history of connective tissue or autoimmune diseases. Her single brother has Crohn’s disease. The patient had no risky sexual behavior or history of substance use and maintained regular contact with a domestic cat. Her symptoms progressed despite antipyretics, and she was admitted to the hospital. She was still in good physical condition and described no gastrointestinal symptoms. The fever ranged between 39 and 40°C without any evident daily pattern. Physical examination revealed painless splenomegaly but no palpable lymphadenopathy and there was no rash, swelling of joints, or synovitis. On presentation, she had mild normocytic and normochromic anemia (Hb 11.7 g/dL), leukopenia (3.1 × 109/L), normal platelet count, C-reactive protein of 97.9 mg/L, elevated lactate dehydrogenase of 415 U/L, hyperferritinemia of 449 ng/mL, erythrocyte sedimentation rate of 31 mm/h, aspartate aminotransferase of 64 U/L, alanine aminotransferase of 31, and mild hypoalbuminemia (32.0 g/L). Other indicators of hepatic and renal function were normal. Chest X-ray was normal. Admission electrocardiography showed sinus tachycardia. A nasopharyngeal aspirate for COVID-19 (real-time polymerase chain reaction assay) and other respiratory viral infections was negative. Blood cultures (aerobes, anaerobes, fungi, mycobacteria), urinalysis, stool samples, pneumococcal, Legionella urinary antigen tests, and interferon gamma (QuantiFERON®) were negative. Epstein-Barr, varicella zoster virus, parvovirus B19, HIV, and hepatitis B and C serologies showed no evidence of active infection. Q fever and rickettsial serologies were negative. Anti-nuclear antibodies, anti-Sm, and anti-dsDNA were all within the reference ranges. A rectosigmoidoscopy was performed and showed no signs of endoscopic activity. Biopsies were taken and immuno-chemistry for herpesvirus and CMV was negative. Fecal calprotectin on day 2 was negative and stool cultures were negative too.
CMV serology detected low IgM and IgG antibody titers, and intravenous ganciclovir (5 mg/Kg twice daily) was started concerning a possible CMV reactivation. Cat scratch disease was deemed as a diagnostic possibility as the patient maintained a regular contact with a cat, so we decided to pursue a therapeutic challenge with doxycycline. However, after 10 days, there was no clear clinical or biochemical response, the quantitative PCR obtained at this time revealed no cytomegaloviremia, and the patient felt sick and feverish. A chest-abdomen-pelvis computed tomography demonstrated sub-mental, submandibular, and bilateral axillary lymphadenopathy, hep-atomegaly, and splenomegaly of 16.5 cm, without an evident focus of infection (Fig.1). Positron emission tomography scan showed hepatosplenomegaly and heterogeneous uptake of 18-FDG diffusely involving the axial and proximal appendicular skeleton, suggesting medullary reactivity. Therefore, there were no functional changes suggestive of high-grade metabolic malignancies or active sarcoidosis. A peripheral blood smear revealed few leucocytes and platelets but no blasts. A bone marrow biopsy demonstrated occasional stromal macrophages but no evidence of hemophagocytosis. Echocardiogram showed normal biventricular systolic function without evidence of endocarditis.
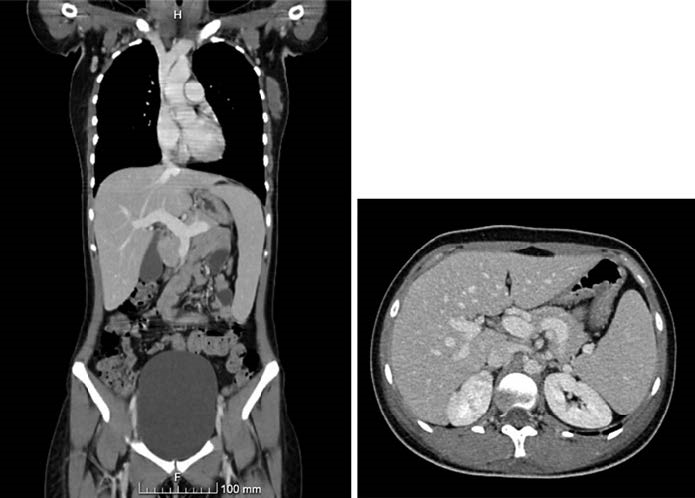
Fig. 1 Chest-abdomen-pelvis computed tomography. a Coronal view. b Axial view. Obvious hepatomegaly and splenomegaly without an evident focus of infection.
Hyperferritinemia of 1,383 ng/mL, soluble CD25 elevated at 2,558 U/mL (negative control 758 U/mL), and fasting triglyceride level elevated at 415 mg/dL (4.69 mmol/L) were noted on the patient’s 15th day of hospitalization. In addition, her liver test values had increased with ALT of 141 U/L, AST of 70 U/L, GGT of 244.4 U/L, and ALP of 52 U/L. A percutaneous hepatic biopsy was performed and showed liver with normal trabecular architecture with multifocal steatosis (Fig. 2).
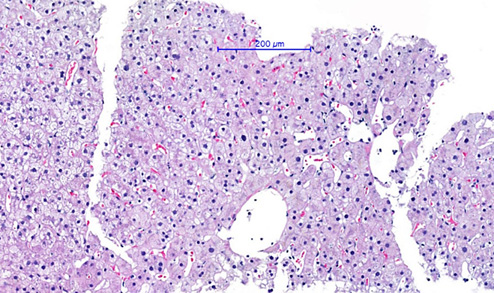
Fig. 2 Liver biopsy findings: normal trabecular architecture with multifocal steatosis but without evidence of bile duct or necro-inflammatory lesions or hemophagocytosis (HE, ×20).
Taking into account the whole clinical picture of high-grade fever, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, soluble CD25 elevation, and bicytopenia in the absence of evidence for infection or malignant disease, a diagnosis of an HLH presumptively secondary to recent COVID-19 vaccination was made. Intravenous dexamethasone (10 mg/m2 daily) was started. She became apyretic 48 h after starting steroids and concurrent symptomatic and biochemical improvements were observed. The patient was discharged home after 30 days to continue her recovery on a weaning course of oral prednisolone. Genetic testing for PRF1, STX11, STXBP2, UNC13D, DCLRE1C, RAG1, and RAG2 was requested, albeit it was not positive for any of these mutations.
Discussion
HLH is a syndrome of hyperinflammation and tissue damage due to irregular immune activation. The dysregulated immune state is thought to be caused by the lack of normal downregulation by natural killer cells and/or cytotoxic T lymphocytes which fail to eliminate activated macrophages [1]. This absence of a normal feedback response leads to disproportionately stimulated benign macrophages in all reticuloendothelial organs such as the spleen, liver, bone marrow, and lymph nodes, with secretion of a huge amount of cytokines like tumor necrosis factor-α (TNF-α), interferon gamma (IFN-γ), interleukin-6 and -10, and macrophage colony-stimulating factor [2]. These pro-inflammatory mediators invade normal tissues, causing histiocytic hemophagocytosis, cytokine storm, and cytokine-mediated biochemical modifications, ultimately resulting in blood vessel destruction and tissue necrosis. Eventually, multiorgan dysfunction and disseminated intravascular coagulation can establish [3], and if not diagnosed and treated promptly, they may be lethal in virtually all cases [4].
Patients with HLH may have a single episode or relapsing episodes, the last ones occurring most frequently in familial HLH [5]. Up to one quarter of HLH cases are thought to be familial [5]. The genetic forms include mutations in CD8+T cells and natural killer cells that result in a loss of performance in the cytolytic pathway proteins (Table 1) and mutations associated with primary immunodeficiency syndromes (Table 2). The acquired form is associated with infections (viruses, bacteria, fungi, and parasites), hematologic and solid malignancies (such as Hodgkin and non-Hodgkin lymphomas), autoimmune diseases (such as systemic lupus erythematosus and seronegative spondyloarthropathies) [9]. Viral infection, particularly Epstein-Barr virus, is the most frequent trigger, either as a primary infection in healthy individuals or after reactivation in patients with a weakened immune system [10].
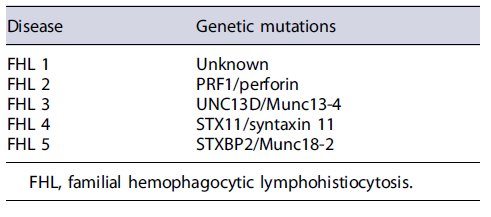
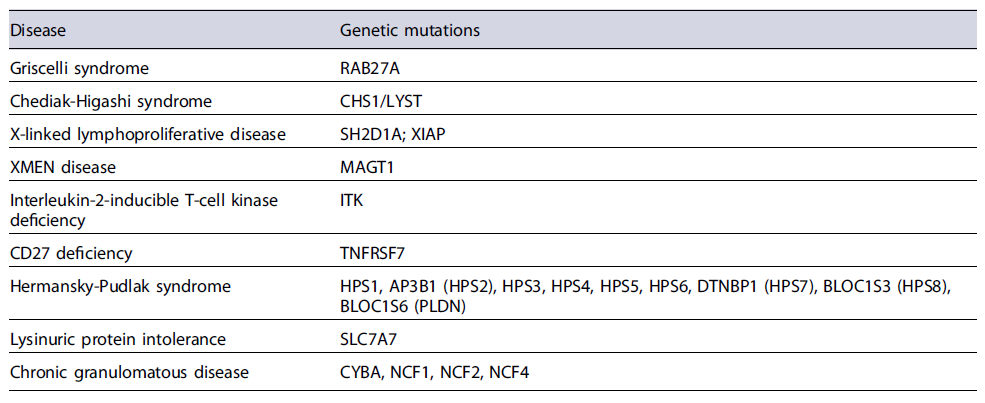
Infants are most affected with the highest incidence in those with less than 3 months. In adulthood, there may be a slight male susceptibility [10, 11], although the incidence rate in adults is not known [2].
The Histiocyte Society proposed clinical and laboratory criteria to help establish the diagnosis of HLH. The diagnosis is established if a patient has a positive genetic mutation or meets at least 5 of 8 criteria [6] (Table 3). A bone marrow biopsy may be performed for patients at high risk or with suspected malignant disease. Genetic testing is indicated in all patients who fulfill the diagnostic criteria for HLH and in those with a high probability of HLH based on the initial evaluation to rule out hereditary HLH mutations.

HLH usually presents as an acute or subacute febrile syndrome associated with hepatosplenomegaly and cytopenias, but none of these characteristics is specific [7]. Some patients may experience recurrent fever of unknown origin and manifest only a subset of the diagnostic criteria.
Typical laboratory results in HLH include high ferritin, triglycerides, transaminases, bilirubin (mostly conjugated), and sCD25 (α-chain of the soluble interleukin-2 receptor) and decreased fibrinogen [7]. A relatively normal ferritin can occasionally be seen in HLH genetic syndromes, even during a disease flare. Hemophagocytosis supports the diagnosis but is neither pathognomonic of nor required for the diagnosis of HLH and can be detected in biopsies of lymph nodes, spleen, liver, or bone marrow aspirates/biopsies. Some patients may show hemophagocytosis only later in the illness course, even if they are clinically improving [7, 8].
Most patients with HLH have hepatitis varying from mildly elevated transaminases to hepatic failure. Liver dysfunction translates into abnormal coagulation with the most frequently reported abnormality being a decrease of fibrinogen levels. Disseminated intravascular coagulopathy is also frequently observed [12]. Increased tumor necrosis factors and chronic inflammation decrease lipoprotein lipase activity, causing high triglyceride levels and cholesterol imbalance [13], but this may not be noticeable until the liver has been affected for some time [14]. Liver biopsy may show an inflammatory hepatitis-like periportal infiltrate in the liver with lymphocytes and histiocytes, explaining the possibility of obstructive jaundice [7].
One-third of patients with HLH may develop neurological alterations such as decreased consciousness, ataxia, seizures, and cranial nerve abnormalities [1, 2]. Other pathophysiological changes of hyperinflammation may be increased capillary permeability with edema and development of acute respiratory distress syndrome, severe hypotension demanding vasopressor support, and renal failure. Skin manifestations such as rashes, petechiae and purpura may also be apparent [3].
The natural history of untreated HLH syndrome is almost uniformly fatal. Prompt recognition of the HLH syndrome and diagnosis of the underlying cause of HLH disease are critical to enabling urgent and appropriate treatment. The fundamental principle of management of HLH includes a short-term strategy to control the hyperinflammatory state, using chemotherapeutic agents, immunosuppressants, and targeted biologic agents, and aims to eliminate activated T cells and macrophages and dampen the cytokine storm. Glucocorticoids are standard, and for patients where cytotoxic therapy is thought to be necessary, the etoposide-based HLH-94 and -2004 protocols and CHOP are commonly used [15]. In patients with genetic-linked HLH, the disorder can be completely eradicated only by hematopoietic stem cell transplantation [16]. Treatment for secondary HLH includes both immunosuppressive therapy and treatment of the underlying disorder, such as infection and malignancy [16]. Survival in HLH is heterogeneous and malignancy/lymphoma-associated HLH is associated with poor outcomes.
Although rare, IBD patients are theoretically at risk of developing HLH, provided the appropriate treatment makes them susceptible to opportunistic infections and malignancies [17]. In fact, opportunistic infection or malignancy was found in more than 80% of all known IBD patients with at least some biologic exposure who developed HLH in a recent systematic review [18]. Studies reporting HLH occurring in IBD show a more frequent occurrence of HLH in patients with Crohn’s disease compared to UC [18, 19], yet the reason behind this susceptibility is not known. In this particular patient, in addition to biologic therapy with vedolizumab, genetic predisposition was another concern raised upon this second episode of HLH, once presentation at a late age, even in adulthood, can occur [20].
The duration between vaccination and the onset of symptoms in this case was in accordance with other reports. The mean duration between diagnosis of underlying HLH trigger and occurrence of first symptoms is around 10 days [9] and probably correlates with the innate cytokine signature triggered within days after COVID-19 vaccination. The exact mechanisms of COVID-19 vaccination-related HLH remain unknown, but it is recognized that immune stimulation by mRNA- or DNA-based vaccines may elicit a massive cytokine storm, which may result in HLH [21]. T-cell activation through antigen stimulation plays a role in HLH pathogenesis and it is likely that T-cell expansion after vaccination could lead to increased IFN-γ production and bystander activation triggering pathologic inflammation [22]. IL1-1β is a pro-inflammatory cytokine largely involved in the hyperinflammation syndrome caused by COVID-19 [23] and its secretion can be stimulated by SARS-CoV-2 spike protein in macrophages. Hence, a potential influence of IL1-1β hyperinflammation syndrome after mRNA SARS-CoV-2 vaccination is a likely mechanism. Molecular mimicry and a potential immune response mediated by anti-spike antibodies have also been implicated [24].
The occurrence of HLH after vaccination is a very rare condition, but it has been described for other vaccines, such as influenza [25, 26], measles [27], and BCG vaccination [28]. There are reports of HLH following other types of COVID-19 vaccines like the ChAdOx1 AstraZeneca® vaccine [29, 30] and inactive vaccine [31]. So far, up to 100 cases of HLH have been temporally associated to COVID-19 vaccination according to Vaccine Adverse Event Reporting System (VAERS) database [32], strengthening the potential relation between COVID-19 vaccination and HLH development. To the best of our knowledge, this is the first case in the literature of HLH after COVID-19 vaccination in a patient with IBD under biological therapy.
With this case, we highlight the need of awareness of the clinical syndrome of HLH to rapidly be able to instigate diagnostic evaluations and early treatment. While the benefits of vaccination are unquestionable and it should be undoubtedly encouraged, close follow-up is needed, especially in individuals with immune dysregulation, as demonstrated in this case.

