











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Lymphoproliferative disorders arising in a background of immune deficiency/dysregulation (primary or acquired) correspond to a spectrum of disorders ranging from non-noxious hyperplasia to aggressive lymphomas1.
In autoimmune disorders, the risk and type of lymphomas seem to be dependent on the immunomodulatory agent used, the dose, duration, and underlying immunosuppressive disorder1.
B-cell lymphomas are the most frequent lymphomas associated with immunodeficiency states, whether primary or secondary1. Hepatosplenic T-cell lymphoma (HSTCL) is the most frequently reported subtype among the T-cell non-Hodgkin lymphomas associated with exposure to immunosuppressive agents1.
We present a case of an atypical cutaneous presentation of a systemic extranodal NK/T-cell lymphoma in a patient with ankylosing spondylitis on long-term therapy with tumor necrosis factor-alpha (TNF-α) inhibitor and prednisone.
Case report
A 60-year-old patient, with a history of ankylosing spondylitis and autoimmune hemolytic anemia, under treatment with infliximab and low-dose prednisone for the past 9 years, was referred to the Hematology Department of our Hospital, for a suspected cutaneous lymphoma. On clinical observation, he presented with a 7 cm ulcerated, well-demarcated lesion on his left lower back and in the left scapular region with a 20 cm erythematous, and scaly patch with two infiltrated 4 cm plaques within it (Fig. 1). On palpation, there were no detectable lymphadenopathies or hepato-splenomegaly. The lesions had a 6-month evolution gradually transitioning from patches to plaques and tumours. A skin biopsy of the tumor phase (and later from the plaque phase), performed in the hospital of origin, was reviewed in our pathology department for a histopathological diagnosis. We observed a diffuse infiltration of the superficial and deep dermis by atypical, intermediate-size lymphocytes, with dispersed chromatin (Fig. 2). The neoplastic lymphocytes, evaluated in paraffin-embedded tissue, expressed CD2, CD3, CD56, TIA-1, Granzyme B, and TCRδ and negativity for CD4, CD5, CD7, CD8, CD20, CD30, BCL6, CD278, TCRβ, and PD-1 (Fig. 3). In situ hybridization analysis with EBER revealed nuclear positivity in all neoplastic cells. There was no evidence of epidermotropism, vasculotropism, or necrosis.
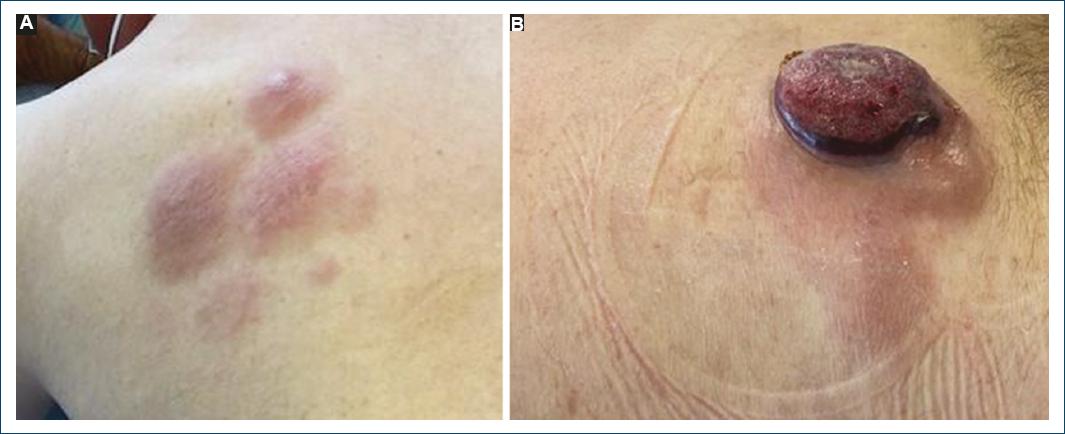
Figure 1 A: patient with a well-demarcated, erythematous, and scaly patch with two infiltrated plaques within it and B: an ulcerated and well-demarcated tumor on his left lower back.
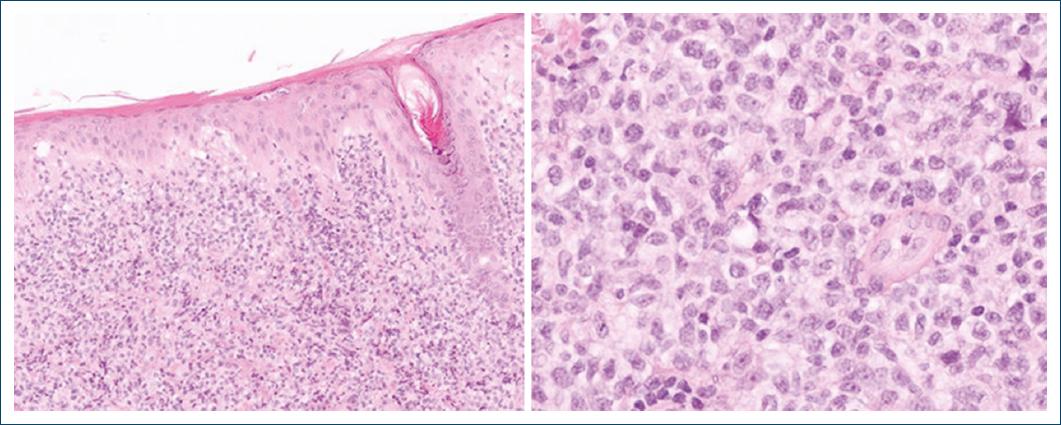
Figure 2 Infiltration of the dermis by atypical, intermediate size lymphocytes, with dispersed chromatin.
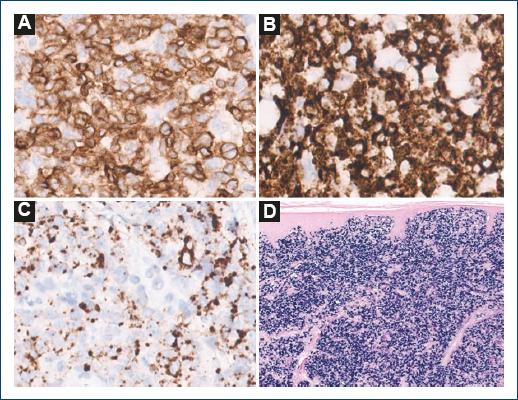
Figure 3 The neoplastic lymphocytes expressed diffuse positivity for A: TCR-δ, B: TIA-1, and C: granzyme B. D: in situ hybridization analysis with EBER revealed nuclear positivity in all neoplastic cells.
Due to the ambiguous features of the case a diagnosis of a γδ T-cell lymphoma, Epstein-Barr virus (EBV)-positive was suggested.
We pursued an investigation to determine whether it was a systemic or a primary cutaneous T-cell lymphoma.
A 2-deoxy-2-[18F] fluor-d-glucose positron emission tomography/computed tomography (FDG-PET/CT) and a bone marrow trephine were performed for disease staging. The FDG-PET/CT revealed a stage IV lymphoma with diffuse hypermetabolism of supra- and infra-diaphragmatic lymph nodes, skin, subcutaneous fat, bone marrow, and homogenous hepatosplenomegaly, with the spleen measuring approximately 16 cm.
The bone marrow trephine revealed a diffuse, mas- sive interstitial infiltration of medium-sized T-cell lym- phocytes, positive for CD3, CD2, CD45, CD56, and TCR ãä by flow cytometry, compatible with infiltration of the bone marrow by a ãä T-cell lymphoma, and identical to the previously described on the skin (Fig. 4). An identical clonal rearrangement of the T-cell receptor delta gene was detected both in the bone marrow and in the biopsy of the skin lesion.
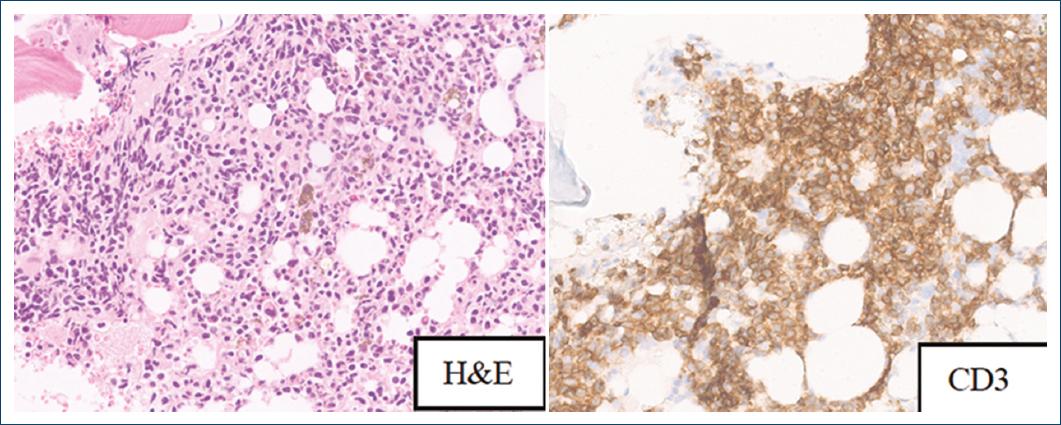
Figure 4 Diffuse interstitial involvement of the bone marrow by CD3-positive neoplastic lymphocytes.
Plasma EBV DNA levels were 88642UI/mL.
In addition, an identical clonal rearrangement of the T-cell receptor delta gene was detected both in the bone marrow and in the biopsy of the skin lesion.
The authors, based on the histological and immunological features of the skin biopsy and bone marrow involvement, together with the systemic nodal and splenic uptake granted a diagnosis of a systemic γδ T-cell lymphoma, EBV-positive, favoring a diagnosis of an extranodal NK/T-cell lymphoma, with cutaneous involvement and probable association with immunomodulating therapy.
The disease proved to be highly refractory to several therapeutic regimens, including cyclophosphamide, doxorubicin, vincristine, prednisolone, rituximab, ifosfamide, carboplatin, etoposide and rituximab and hyperfractioned-cyclophosphamide, vincristine, doxorubicin, and dexamethasone. In addition to the refractoriness of the disease, several serious infectious complications were documented, as well as hematologic and gastrointestinal toxicity. Following this series of events, the patient died, 2 months after the initial diagnosis (Table 1).
Table 1 Clinicopathologic features of extranodal NK/T-cell lymphoma in a patient with ankylosing spondylitis receiving TNF-α inhibitors
| Age/gender | Associated immune disease | Involved organs | Immunomodulator | IHQ-panel | EBV status* | Treatment | Survival+ | Status |
|---|---|---|---|---|---|---|---|---|
| 60 M | Ankylosing spondylitis | Skin, liver, spleen, bone marrow, supra and infra diaphragmatic lymph nodes | Infliximab + prednisone | CD2+, CD3+, CD4-, CD5-, CD7-, CD8-, CD30-, CD56+, TIA-1+, Granzyme-B+, TCR-d+, TCR-Β-. | +† | CHOP R-ICE HYPER-CVAD | 2 mo | Passed away |
*EBV status determined by in situ hybridization with EBER probe.
†Nuclear positivity in all neoplastic cells. Survival+ represents time, in months (mo), from diagnosis. R-ICE: rituximab, ifosfamide, carboplatin, etoposide.
Discussion
γδ T lymphocytes are tissue-restricted cytotoxic lymphocytes accounting for < 5% of the adult T-cell population. They are crucial in immunosurveillance and are mainly lodged in the skin, sinusoids of the liver, red pulp of the spleen, and the intestinal mucosa2.
Persistent, dose-dependent, treatment with infliximab has been shown to promote the expansion of clonal γδ T-cells in vivo and induce proliferation in vitro3. Furthermore, patients with inflammatory bowel disease or psoriasis seem to have a higher baseline frequency of γδ T-cells when compared to the general population, and, when treated with thiopurines or methotrexate in association with infliximab, demonstrate an even higher baseline frequency and a lower threshold for their expansion3.
In the literature, so far, seven cases of T-cell lymphomas in association with TNF-α inhibitors have been documented, in patients with ankylosing spondylitis4-8. Curiously, the predominant histological subtype is Mycosis fungoides/Sezary syndrome. Other types include subcutaneous panniculitis-like TCL, HSTCL, angioimmunoblastic T-cell lymphoma, and one case of anaplastic large-cell lymphoma, also with a cutaneous presentation4-9.
The description of extranodal NK/T-cell lymphomas, in patients on infliximab therapy, is extremely rare10-12.
In our case, the cutaneous manifestation of extranodal NK/T-cell lymphoma presented after a 9-year immunosuppression treatment, with infliximab and low-dose prednisone, for ankylosing spondylitis. The systemic presentation, with nodal involvement, also favors this diagnosis over other γδ T-cell lymphomas, such as HSTCL.
This case represents a diagnostic dilemma with clinical, morphological, immunohistochemical, and prognostic overlap to other γδ T-cell lymphomas.
The differential diagnosis includes of a cutaneous γδ cytotoxic T-cell lymphoma is included extranodal NK/T-cell lymphoma (by definition EBV-positive), primary cutaneous γδ T-cell lymphoma (by definition EBV-negative), and cutaneous involvement of an HSTCL (by definition EBV-negative).
In this case, an angiodestructive growth pattern and necrosis, common and desirable features of an extranodal NK/T-cell lymphoma, were not observed1. Furthermore, involvement of the bone marrow is rare in extranodal NK/T-cell lymphoma. On the other hand, HSTCL is an EBV-negative lymphoma, although rare reports of EBV positivity, as a secondary event of reactivation of the virus, have been reported1. HSTCL usually presents with an intrasinusoidal infiltration of the bone marrow and absence of nodal disease1. Rare EBV positivity has also been described in primary cutaneous γδ T-cell lymphoma2, but a systemic disease at diagnosis is extremely rare for a primary cutaneous γδ T-cell lymphoma13.
For all the above, the histological diagnosis of this case is particularly challenging and emphasizes the importance of a complete clinical history, including past or current medication, and illustrates the difficulty of adequately classify EBV-positive γδ T-cell lymphomas according to the current standardize classification of haematolymphoid tumors. We reinforce the need to further expand and review the classification of these entities.
The cautious use of immunomodulators, including TNF-α inhibitors, high clinical suspicion, and surveillance of possible consequences of prolonged immunosuppression, is warranted to avoid a rapid fatal disclosure associated with the risk of an aggressive lymphoma14,15.
Conclusion
The histological diagnosis of this case is particularly challenging and emphasizes the importance of a complete clinical history, including past or current medication, and illustrates the difficulty of adequately classify EBV-positive© T-cell lymphomas according to the current standardize classification of haematolymphoid tumors. We reinforce the need to further expand and review the classification of these entities.
The cautious use of immunomodulators, including TNF inhibitors, high clinical suspicion, and surveillance of possible consequences of prolonged immunosuppression, is warranted to avoid a rapid fatal disclosure associated with the risk of an aggressive lymphoma14,15.

