











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Case Presentation
A 78-year-old female patient was admitted to the emergency department of our institution with a history of psychomotor retardation, myoclonus and dysarthria, over the last 2 months. Her past medical history only included atrial fibrillation with a rapid ventricular response. The neurological examination confirmed the findings mentioned above and an electroencephalography was performed which showed the presence of encephalopathy. The patient was admitted with a presumptive diagnosis of transmissible spongiform encephalopathy (Creutzfeldt-Jakob disease). A cerebral MRI (not shown) was conducted, which not only excluded this diagnosis but also revealed the presence of areas of hyperintense signal on T1-weighted images with ill-defined margins, symmetrically affecting the globus pallidus and the medial region of the cerebral peduncles’ feet. These findings were compatible with liver encephalopathy. For this reason, a Computed Tomography (CT) scan of the abdomen was performed and showed a large intrahepatic portosystemic shunt between the left branch of the portal vein and the left hepatic vein in a non-cirrhotic liver (Figure 1A - 1C).
In addition, a liver biopsy was performed which showed normal architecture with no sign of inflammation or fibrosis.
The diagnosis of encephalopathy associated with portosystemic shunt bypass without intrinsic hepatocellular disease was made.
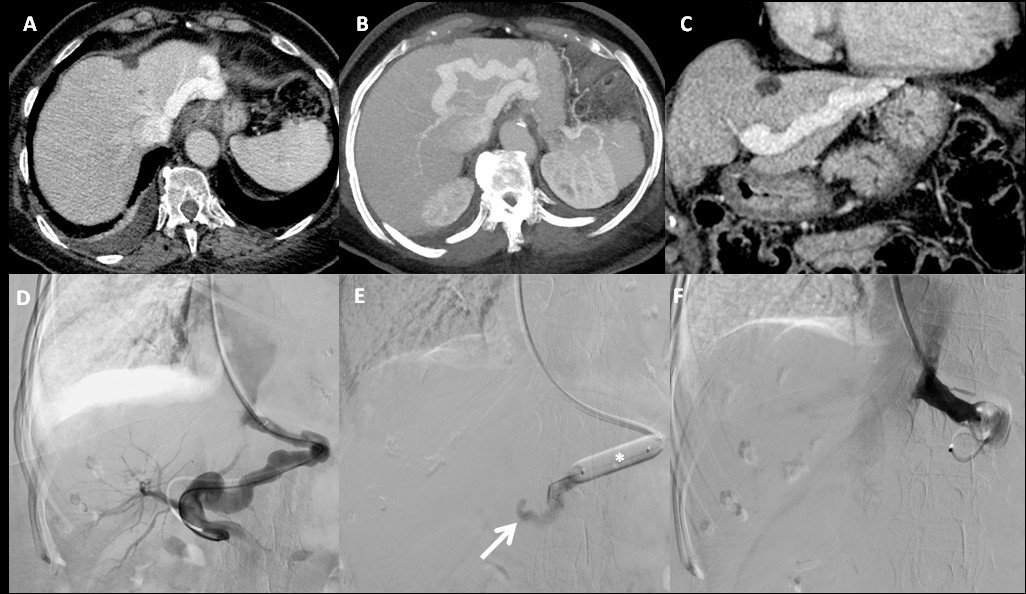
Figure 1: A - Axial portal phase CT, 3 mm slice thickness; B - Axial portal phase CT, MIP reformation = 15 mm slice thickness; C - Coronal portal phase CT, 3 mm slice thickness; D - Abdominal venography; E - Balloon shunt occlusion test; F - One-step closure of the shunt with two 20 mm Amplatzer plugs.
Digital Subtraction Venography (DSV) was performed. The intrahepatic portosystemic shunt between the left branch of the portal vein and the left hepatic vein was catheterized via a transjugular route (Figure 1D) and then a balloon (*) shunt occlusion test (Figure 1E) was performed, which revealed an adequate left portal vein (arrow) and an increase in portal pressure of only 2 mmHg with shunt occlusion. Since there was no excessive rise in portal pressure, the portosystemic shunt was subjected to a one-step closure with two 20 mm Amplatzer plugs (Figure 1F).
The patient made an uneventful recovery. She has had no episodes of encephalopathy in a follow-up period of 1 year.
Encephalopathy associated with portosystemic shunt bypass without intrinsic hepatocellular disease is often referred to as Type B hepatic encephalopathy as illustrated in this case with ammonia being the mediator of the neuropsychiatric manifestations.1 Under normal conditions, ammonia is metabolized to urea by the urea cycle in the liver but in the presence of a portosystemic shunt, ammonia bypasses hepatic metabolism and entering the systemic circulation and further transitioning through the blood-brain barrier.2
Congenital portosystemic shunts (CPSS) are rare vascular malformations that consist in abnormal connection(s) between any vein of the portal system and any systemic vein especially of the inferior vena cava (IVC) system or the right atrium. They result from the incomplete involution of one or several early embryonic or fetal vessel(s) during fetal life or after birth for the ductus venosus (DV).3
CPSS can cause partial or complete diversion of the portal blood to the systemic vessel and carries a risk of severe multisystemic complications. They differ from acquired portosystemic shunt occurring as a consequence of portal hypertension.3
Broadly, CPSS have been classified as intrahepatic and extrahepatic.4 Park et al. proposed a classification for intrahepatic CPSS, dividing them into subtypes based on location, number and shunt characteristics.4 There are four subtypes of intrahepatic CPSS. The most common is type 1 in which a solitary large vessel connects the right portal vein with the IVC.5 The second most common is type 2 in which the distal part of the portal vein abnormally joins branches of the hepatic vein specifically within one hepatic lobe. In type 3 the shunt is arising from a congenital aneurysm between the peripheral branches of the portal vein and the hepatic vein. Finally, in type 4 multiple anastomoses occur between branches of both venous systems involving both hepatic lobes.6
Despite the type, CPSS are typically found incidentally. However, they may be associated with other congenital abnormalities such as duodenal atresia, absence of the DV, shunts associated with other organs, etc. Clinical presentation depends on the degree and the type of shunt, ranging from an incidental finding on imaging to hepatic encephalopathy.7
There are many complications associated with these anomalies including pulmonary congestion leading to pulmonary hypertension in which patients will usually present with gradual shortness of breath, hepatorenal syndrome characterized by gradual worsening of renal function and signs of fluid retention, and acute manifestations similar to the case of our patient presenting with altered sensorium and hepatic encephalopathy. The prognosis of the disease depends on the extent of cardiac, renal and hepatic complications. Patients like in this case, in which the condition is limited to only the hepatic system have a better prognosis.8
As the clinical presentation is nonspecific, imaging is critical for the diagnosis of CPSS.
Ultrasonography with Doppler study is usually the first-line exam that shows abnormal communication between the portal and the systemic vein and the consequences on liver morphology, the presence of liver nodules, and the association with other malformations. It is also very useful for the follow-up of CPSS before and after treatment.9
CT and MR imaging are helpful to confirm the diagnosis, to provide a precise vascular map and to search for complications and associated malformations.3
Treatment of CPSS varies and depends on the severity of clinical sequelae. Several may close spontaneously; however, when the CPSS remains patent, radiologic or surgical closure of the CPSS may prevent, resolve, or stabilize complications.3
Endovascular closure is the first option for treatment when possible, because it is the least invasive method and results in rapid amelioration of symptoms, correction of high ammonia levels, and regression of liver lesions.10,11
Embolization of the shunt using various materials including coils and microcoils, detachable balloons, and N-butyl cyanoacrylate lipiodol mixtures or recently introduced multilayer devices (vascular plugs) has been reported depending on shunt size.12,13During the procedure, an occlusion test with a temporary balloon placement is recommended to record portal vein pressure elevation. If the portal pressure exceeds 30 mmHg, then permanent occlusion at this stage should be avoided, as it is likely to cause significant changes in liver hemodynamics, derangement of liver function, and potential worsening of preexisting symptoms and gastrointestinal bleeding. A two-stage approach is advocated for these cases with the use of a size reduction stent followed by definitive occlusion (radiological or surgical ligation) a few months later and will allow the liver to compensate and the portal pressure to reduce gradually without complication.14,15,16
Other therapeutic options may include surgical ligation or, rarely, liver transplantation.3
In summary, vascular malformation as a cause of unexplained encephalopathy should always be considered especially in patients with no underling liver cirrhosis, as represented in this case. CPSS are rare and represent persistent embryologic connection between portal system and the systemic veins. Early recognition and correction with either radiological or surgical occlusion reverses symptoms and prevents long-term complications.

