











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder of DNA repair with 100% penetrance and an estimated incidence in the United States and Western Europe of approximately 1 per 1,000,000 live births [1]. XP affects, mostly, sun-exposed skin and eyes due to an increased sensitivity to ultraviolet radiation (UVR). Mutations in any of the eight genes involved in the recognition and repair of UVR-induced DNA damage can be encountered, which ultimately lead to eight different subgroups: XPA through XPG, and XP-V. Both genders are equally affected. Compared to the general population, individuals with XP are at significantly higher risk of developing skin cancer, presenting at an earlier age. A clinical diagnosis is based on typical skin findings, including freckling before the age of two, xerosis, progressive atrophy and hyper/hypopigmentation. Severe solar erythema also occurs after minimal exposure to radiation, although this feature is not usually perceived in XPC, XPE and XP-V subgroups, whose reaction to sun exposure is globally normal [2]. Ocular findings such as photophobia, xerophthalmia, conjunctival injection, ectropion and keratitis are also found [3], and, less commonly, neurodegenerative diseases, including sensorineural hearing loss, ataxia, areflexia, and peripheral neuropathy [4]. Along with clinical findings, a positive family history can support the diagnosis. Genetic testing confirms and categorizes the patient into one of the XP subgroups. In these patients, it is strictly recommended regular skin and eye examination, as well as avoiding sun exposure [5], to ensure an early management of premalignant and malignant skin lesions.
Hereditary hemochromatosis (HH) is an autosomal recessive disorder, widely identified in Caucasians, in which, typically, biallelic inheritance of the p.C282Y variant in the HFE gene leads to inappropriately low levels of hepcidin which, by facilitating ferroportin mediated iron export, results in greater intestinal iron absorption with subsequent iron overload and tissue damage over time[6]. It takes about four decades in males and five to six decades in females to reach the iron threshold (20g) associated with severe clinical manifestations [7]. Homozygosity for the p.C282Y variant (C282Y/C282Y) has an approximate prevalence of 1/150 to 1/300 in populations of northern European descent [8].
A genetic test with HFE genotyping is warranted in patients with hyperferritinemia and high transferrin saturation, confirmed in two determinations, after other frequent causes of hyperferritinemia have been excluded. Phlebotomy is the treatment of choice to achieve ferritin targets between 50-100 µg/L and preventing the complications associated with tissue iron deposition. First-degree family members must be screened with ferritin, transferrin saturation and HFE genotype analysis[9].
We report a case of a 51-year-old woman, with a medical history of XP, who has been diagnosed with HH. After genetic testing, it was possible to infer a possible linkage between two mutated neighbor genes (HFE and POLH).
Case Report
A 51-year-old Caucasian woman, with a history of skin hyperpigmentation, mainly in sun-exposed areas, since the age of 4 was clinically diagnosed with XP at 27 years. She has been previously submitted to complete excision of three basal cell carcinomas and two keratoacanthomas.
The patient was referred to our Hepatology Department, due to upper quadrant abdominal pain with evidence of liver enzymes abnormalities and an abdominal ultrasound showing loss of structural homogeneity, hypertrophy of the caudate lobe and mild splenomegaly, all suggestive of chronic liver disease. In addition to the XP diagnosis, the patient’s medical history was notable for a history of menopause at the age of 40, essential arterial hypertension, osteoporosis, alcohol consumption of 12 g/day and no other risk factors for liver disease, namely, drugs or sexual risk behaviors. She was unaware of family history of hereditary diseases or consanguinity. Her physical examination was remarkable for a good general condition, a body mass index of 22.19 kg/m2, a waist circumference of 89 cm and marked skin hyperpigmentation (Fig. 1). The initial blood testing found abnormalities on liver enzymes (AST 84 U/L; ALT 75 U/L; ALP 106 U/L; GGT 286 U/L), thrombocytopenia (platelets 89 ×103/μL), elevated transferrin saturation (90%) and ferritin levels (4,806 ng/mL) (Table 1). An esophagogastroduodenoscopy was performed, showing no signs of portal hypertension.
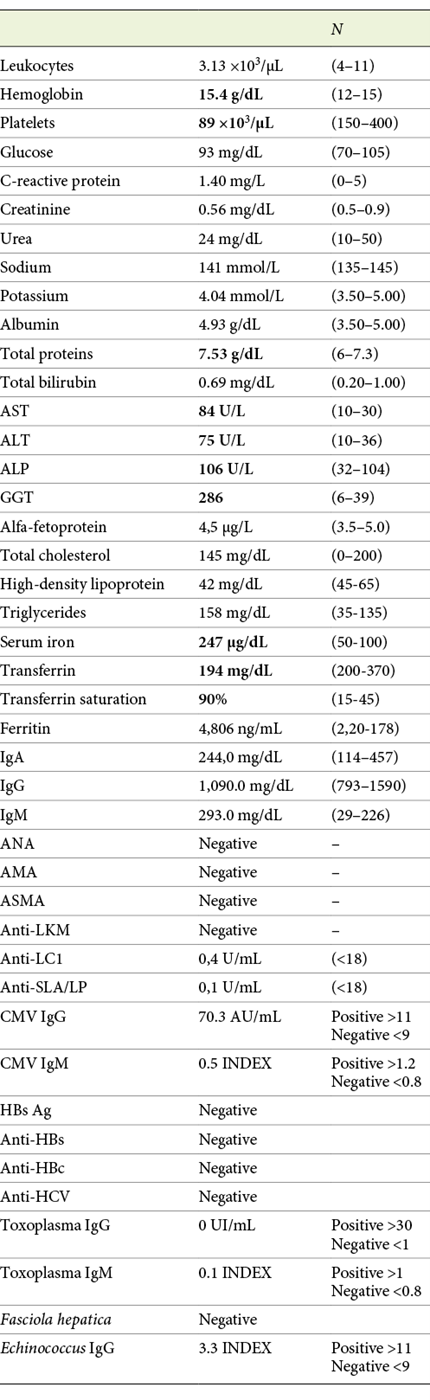
Fig. 1: Multiple solar lentigines and xerotic appearance in the photo-exposed area. Note the clear demarcation with the zone usually protected by clothes.
Due to the substantial increase in ferritin and transferrin saturation levels, HH diagnosis was considered and later confirmed after HFE genotyping revealed C282Y homozygosity.
Additionally, magnetic resonance imaging was requested (Fig. 2), which showed a severe iron overload and estimated hepatic iron concentration of 300 micromoles per gram of dry weight (normal <36). Liver biopsy documented complete architectural disorganization and presence of regenerative nodules, confirming the diagnosis of liver cirrhosis, abundant hemosiderosis and severe steatosis (Fig. 3).
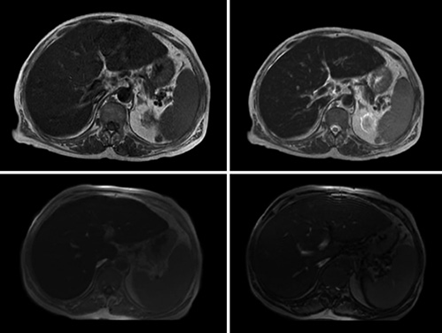
Fig. 2: MRI documenting in axial T1-weighted (upper left) and T2-weighted (upper right) images a decrease in the liver signal intensity. Axial T1-weighted gradient echo sequence images (lower panel) show a decreased signal intensity in the liver on the in-phase image (left) compared with the out-of-phase image (right), corroborating hepatic iron overload.

Fig. 3: Fragment of liver biopsy with hematoxylin-eosin staining (100×), in which iron pigment stains brown (on the left); fragment of liver biopsy with Pearlsstaining (100×), in which the iron pigment stains blue (on the right).
Dietary care was explained, focusing on alcohol cessation, and weekly therapeutic phlebotomies were started. Soon after, the patient developed large volume ascites, managed successfully with hyposaline diet and diuretics, without recurrence. Target values of ferritin were reached in 68 weeks, as well as normalization of serum transaminases, corresponding to an estimated amount of 7.9 g of iron removed. The patient started maintenance therapy with phlebotomies every 2 months thereafter. Additionally, she was administered with the Hepatitis B and pneumococcal vaccines and recommended the Influenza vaccine annually. Family screening was carried out and C282Y homozygosity was detected in two siblings and one grandson.
After a 10-year follow-up, the patient remains asymptomatic, with unremarkable blood tests. Liver ultrasound shows only evidence of diffuse steatosis. During this time interval, she was diagnosed with actinic keratosis on the right ala of the nose and on the right forearm, managed with cryotherapy in the Dermatology Department. Due to the known proximity of HFE (6p22.2) and POLH (6p21.1) genes, the patient was offered a genetic test to detect the latter mutation and was found to carry a rare, but potentially deleterious, homozygous variant of the POLH gene c.571A>C (p.(Thr191Pro)), of uncertain significance.
Discussion/Conclusion
This patient presented with laboratory and imaging evidence of chronic liver disease, including increased liver transaminases, GGT and ALP. The deposition of iron in hepatocytes does not cause inflammation per se[7] and most patients with hemochromatosis present transaminases within normal values. Given our patient history, it was not possible to exclude the role of alcohol consumption or metabolic syndrome as possible contributing factors for this elevation. In fact, excess alcohol intake is a major risk factor for the development of liver disease in patients with HH and iron overload potentiates the development of alcoholic liver disease [10]. Ferritin and transferrin saturation levels largely exceeded established cutoff values in our patient. Since ferritin is an acute-phase protein, it may also be increased in the context of systemic inflammatory processes, but usually at lower values and without increased hepatic iron storage [7].
Given that a definite diagnosis of HH can be made in the presence of p.C282Y homozygosity and increased iron stores, the patient was offered a genetic test, which confirmed HH. C282Y/C282Y has a 13.5% penetrance and is mainly associated with liver abnormalities, namely liver fibrosis and cirrhosis in 5 and 2% of women, respectively [11, 12]. Although the diagnosis of menopause is clinical, determination of serum levels of FSH and estradiol can be useful to confirm the diagnosis in women aged 40-45 years [13]. In this woman, it would have been imperative to rule out the contribution of hypogonadotropic hypogonadism as a cause of secondary amenorrhea, which may have been misinterpreted, at first, as early menopause. Osteoporosis may be a direct consequence of hypoestrogenism, which leads to increased bone resorption.
Liver biopsy was the gold standard for the diagnosis of HH before HFE genotyping became widely available. Although it is not necessary for the diagnosis of HH, it can be offered to C282Y homozygous individuals with ferritin levels >1,000 ng/mL, for the purpose of determining the presence or absence of advanced fibrosis or cirrhosis. Also, where there is hyperferritinemia with confounding cofactors, such as alcohol consumption or metabolic syndrome, like in our patient, liver biopsy may still be necessary to determine whether iron stores are increased or not [9, 14]. The histological pattern favored the diagnosis of liver cirrhosis, with very severe hemosiderosis, supporting the clinical diagnosis of hemochromatosis.
Following the HH diagnosis, phlebotomies were initiated, reaching the target values of ferritin at the end of 68 weeks and normalization of serum transaminases. Since an improvement in biochemical parameters was achieved, as well as non-recurrence of ascites after ferritin targets were attained, we may assume that iron removal, along with cessation of alcohol consumption, contributed to the decrease in serum transaminases and eventually could represent a regression of liver fibrosis. Although regression of liver fibrosis is described to occur in about 15-50% of HH patients [15], it is not known if reversal occurs in established liver cirrhosis, which is widely accepted as a poor prognosis and worse survival factor [16].
Although this woman had not been genetically tested for XP before, the clinical findings were supportive of an XP-V subgroup, since there was no abnormal skin reaction to sun exposure. This feature might have contributed to poor adherence to prophylactic measures, which led to hyperpigmentation and early malignant skin cancers. XP-V has been found to be caused by POLHgene mutations, which prevent the production of any detectable DNA polymerase eta. A loss of this enzyme prevents cells from replicating UVR-damaged DNA effectively [17].
Due to the proximity of POLH and HFE genes, which can both be found in the short (p) arm of chromosome 6 at position 21.1 [17] and 22.2 [18], respectively, we suppose they could be linked, resulting in their inheritance as a unit, instead of assorting independently during meiosis. Therefore, the patient was offered a genetic test, which evidenced homozygosity for a rare sequence variant (c.571A>C) in the POLH gene. This mutation is of uncertain significance, so it does not confirm nor excludes the diagnosis of XP. However, considering the phenotype presented by our patient, this variant is most likely accountable for XP.
The most similar case was reported in 1932, concerning a 60-year-old cirrhotic patient who had mottled pigmentation of the extremities and face. Although interpreted as hemochromatosis at first, a specimen from a pigmented macule on the hand revealed a typical histologic pattern of XP [19]. Despite the diagnosis of hemochromatosis was not put forward by the authors, we believe this case could correspond to the first association of both diseases.
This case highlights, overall, the importance of considering associated diseases when evaluating our patients. Despite being rare, both diseases have effective, prognostic changing interventions, such as avoiding sun exposure and regular skin examination to prevent skin cancers, in XP, and regular phlebotomies for HH, to avoid deleterious tissue deposition of iron and prevent, among others, liver cirrhosis. Our patient presented skin hyperpigmentation, worsening over time, and initially attributed to the XP, which led to a delay of HH diagnosis, already in a cirrhotic stage.
In conclusion, we report the first confirmed case, to our knowledge, of a patient diagnosed both with XP and HH, in whom two mutated neighbor genes - POLH and HFE - were identified. We believe this was not a random association but is possibly the result of genetic linkage. Since both conditions may present with overlapping clinical features, as skin hyperpigmentation, we pretend to alert clinicians to this possible association, since timely diagnosis of both XP and HH allows early guided interventions and improves patient outcomes.

