











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Digestive hamartomatous polyps may be solitary or multiple, the latter often associated with genetic predisposition [1, 2]. Solitary Peutz-Jeghers (PJ)-type hamartomatous polyps represent a rare and distinct entity from the classic PJ syndrome, an autosomal dominant genetic disorder characterized by the development of multiple polyps in the gastrointestinal (GI) tract in association with patches of hyperpigmentation in the mouth, hands and feet [1, 3]. It is important to distinguish these two entities since the latter is associated with a lifetime cumulative risk of up to 93% for development of malignancies (such as colorectal, breast, small bowel, gastric and pancreatic cancers), but the first seems benign in its course. Solitary PJ polyps are diagnosed in patients with an isolated hamartomatous polyp of the GI tract, no familiar history of polyposis and no typical phenotype [3].
An 80-year-old man with a history of sigmoidectomy due to an obstructive T2N0M0 colorectal cancer under-went a surveillance thoracoabdominopelvic CT scan showing a voluminous endoluminal polyp at the caecum (Fig. 1).
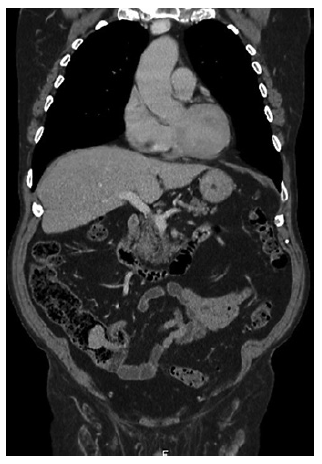
Fig. 1 Thoracoabdominopelvic CT scan (coronal section) with intravenous contrast showing a voluminous endoluminal polyp at the caecum measuring 40 × 31 mm with homogeneous enhancing. No alterations at surgical anastomosis level.
He was referred to our reference centre for colonoscopy. A 4-cm diameter pedunculated and congestive polyp was identified with a short and thick stalk arising from the appendiceal orifice. We proceeded to its en bloc resection using a 25-mm oval diathermic snare after stalk injection with diluted adrenaline 1:10,000, normal saline and methylene blue with polyp recovery for histology (Fig. 2a-c, 3a). The polyp was confirmed to be a hamartomatous polyp of the PJ type (R0 resection) with arborizing pattern of vascularized smooth-muscle tissue axes covered by elongated veliform crypts and occasional in-traluminal necrosis with no dysplasia (Fig. 3b). The patient had no typical manifestations of PJ syndrome or family history. His follow-up showed no complications related to the procedure, including bleeding or acute ap-pendicitis (prophylactic antibiotic was not used).

Fig. 2 a, b Colonoscopy performed showing a 4-cm diameter pedunculated and congestive polyp with a short and thick stalk arising from the appendiceal orifice. c En bloc endoscopic resection was performed using a diathermic snare after stalk injection with diluted adrenaline 1:10,000, normal saline and methylene blue with no complications.

Fig. 3 a Macroscopic specimen of endoscopic resection. b Histopathological analysis revealed a hamartomatous polyp of Peutz-Jeghers-type with arborizing pattern of vascularized smooth-muscle tissue axes covered by elongated crypts with veliform pattern and occasional intraluminal necrosis with no dysplasia (R0 resection).
Solitary PJ-type hamartomatous polyps of the GI tract are rare, and appendiceal location is even rarer. According to the largest case study to date on the follow-up of solitary PJ polyps, they are normally large in size (>15 mm), mostly pedunculated and localized primarily in the colon (even though they have been described in all of the GI tract, except in the oesophagus) [3]. During a mean endoscopic follow-up of 4.5 years (range: 0.1-16.1 years) after excision, no recurrence was observed, making endoscopic follow-up after diagnosis of a PJ polyp unnecessary [3]. Although malignant transformation seems rare, extensive genetic and epigenetic changes have been described in this type of polyps that may contribute to cancer risk [4]. Adding to the risk of other complications related to intraluminal polyp growth (colonic obstruction, intussusception, appendicitis or GI bleeding), and since endoscopic resection allows for a histological diagnosis, polypectomy is always advisable [2, 5]. This case highlights the importance of recognizing the existence of solitary PJ polyps as a distinct entity from PJ syndrome, with drastic consequences of misdiagnosis in terms of follow-up and prognosis. Additionally, the appendiceal location of these polyps is rare and represents a very technically challenging location for endoscopic therapy.

