











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Drug-induced lupus erythematosus (Di-LE) is an autoimmune phenomenon1 that can affect the skin and/or multiple body systems, with a phenotype typically similar to idiopathic lupus erythematosus (LE). It occurs after chronic exposure to a particular drug (usually over months or years of use) and tends to resolve after drug discontinuation2. This entity has been gaining more and more relevance in clinical practice, currently considered to represent approximately 15% of all causes of LE3.
As with idiopathic LE, Di-LE can be classified into drug-induced systemic LE (Di-SLE) and drug-induced cutaneous LE (Di-CLE), presenting either as the subacute or chronic subtype4. The differential diagnosis between drug-induced and idiopathic cases can be a challenge since clinical aspects, serology, and histopathology are identical5. However, they tend to differ in the extent to which they involve different organs and in their clinical course, since Di-LE usually presents as a milder clinical picture with fewer complications2.
Drugs can either unmask clinically silent LE, induce LE exacerbations in a patient that has already been diagnosed (as reported with abatacept6), or trigger a “lupus-like” syndrome, which is the most frequent case2.
There is difficulty in diagnosing this entity due to the lack of validated criteria, but it is important to draw attention to the relevance of timely identification of Di-LE and suspension of the culprit drug, which may allow disease remission. With a low awareness of this condition, the aim of this review is to systematize current knowledge about pathophysiology, clinical, and serological disease manifestations, with an update of associated drugs.
Epidemiology
Di-LE may account for approximately 15% of all LE cases3. It occurs mainly between 55 and 60 years of age7, mostly in females and Caucasians8, but there are also rare pediatric cases reported in patients under treatment with infliximab, carbamazepine4, and valproic acid9.
Pathogenesis of drug-induced LE
The pathogenesis of Di-LE remains poorly understood. The fact that several drugs with distinct chemical structures and different pharmacological actions may be associated with Di-LE contributes to the hypothesis that multiple mechanisms are involved, and, in some cases, they may coexist2. Genetic susceptibility, drug biotransformation, and epigenetic dysregulation, with changes in innate and adaptive immune response seem to be involved4.
Given the usual rapid clinical improvement after drug discontinuation, the autoimmune response in Di-LE can be considered as a transient change in immune response and not a significant affectation of the immune tolerance as in idiopathic LE2. The risk of a drug to induce Di-LE increases with the number of changes that it causes in the individual's immunity. Procainamide and hydralazine, two of the drugs most frequently associated with Di-LE, induce changes both in the innate and adaptive immune response2.
Genetic susceptibility
Genetic susceptibility is evident in Di-LE, but risk factors are different for each drug. Human leukocyte antigens (HLA) DR2, DR3, DR42, and HLA-B83 have been associated with an increased risk for Di-LE induced by minocycline, terbinafine, and hydralazine2. Hereditary complement deficiencies, namely, C4 null allele2 and selective immunoglobulin A (IgA) deficiency, particularly with concomitant HLA-B8 and DR3 haplotypes, have also been hypothesized as risk factor for Di-LE10.
The slow acetylation phenotype may be a risk factor as some drugs inducing Di-LE, such as procainamide and hydralazine, are metabolized by acetylation through the enzyme N-acetyltransferase4 and, therefore, slow acetylators may accumulate more antibody-inducing metabolites2.
A family history of SLE or Di-LE11 or a personal history of another connective tissue disease12 may also be considered a risk factor, as reported for terbinafine3.
Epigenetic dysregulation and autoreactivity/loss of tolerance
Biotransformed drugs and some of their metabolites are responsible for altering the epigenetic properties of B- and T-cells, leading to the formation of autoreactive cells that can induce Di-LE4.
Both hydralazine and procainamide inhibit DNA methylation in T-cells by decreasing the activity of DNA methyltransferase-14 and hypomethylation of T-cell DNA which can alter gene expression profiles and, consequently, T-cell function8. This also results in increased expression of lymphocyte function-associated antigen 1 (LFA-1), leading to increased T-cell reactivity and loss of peripheral tolerance4, which may also occur in idiopathic SLE2.
In addition, reactive metabolites of procainamide and hydralazine can interfere with central T-cell tolerance, leading to the production of autoreactive T-cells, with hydralazine leading B-cells to produce anti-histone antibodies (H2A-H2B-DNA)4.
Drug-induced alterations in innate and adaptive immunity
Drugs and/or their reactive metabolites can activate several pathways within the innate immune response and, therefore, enhance the presentation of self-peptides inducing autoimmunity or they can function as haptens and bind to macromolecules triggering an immune response with activation of autoreactive T and B lymphocytes, for example, by antigen mimicry4. Given the time lag between drug exposure and onset of clinical and serological abnormalities, the biotransformation of the drug into reactive metabolites is probably responsible for autoimmunity, rather than the drug itself1.
Inhibition of the classical complement pathway can also contribute to the pathogenesis of the Di-LE, as in the case of hydralazine, penicillamine, isoniazid, and metabolic products of procainamide4. These drugs can inhibit the covalent binding of complement factor C4 (classical pathway), increasing the concentration of circulating immune complexes by decreasing their clearance4.
Quinidine and procainamide inhibit the removal of apoptotic cells by macrophages which allow a greater number of self-antigens to remain longer in circulation and enhance autoantibody formation4.
Neutrophil extracellular traps (NETs), formed on neutrophil death/apoptosis and consisting of extrusion of a “network” of nuclear DNA and cytosolic proteins, have an important role in host defense, but increased NET formation (NETosis) and/or decreased NET clearance has been associated with different autoimmune diseases, including Di-LE2. NETs function as a source of nuclear material rich in autoantigens and granule proteins that enhance the formation of autoantibodies or autoreactive T-cells4. In addition, they can cause direct toxicity in host tissues, especially in blood vessels2.
Both hydralazine and procainamide promote NETosis, the first by increasing calcium influx and activation of peptidyl arginine-deiminase-4 that mediates chromatin decondensation13 and the latter by activating the muscarinic receptors of neutrophils4 and propylthiouracil increases the production and decreases the clearance of NETs2. However, other Di-LE-inducing drugs such as minocycline and clozapine do not lead to NET formation2.
Procainamide oxidation by activated neutrophils produces hydroxylamine (PAHA, a toxic metabolite) that combines with neutrophil myeloperoxidase (MPO) and creates cytotoxicity4 and hydralazine binds to MPO in intracytoplasmic neutrophil granules enhancing the release of cytotoxic and cell death products13,14. This type of MPO-induced cytotoxicity enhanced by drug-causing LE in vivo is related to their ability to serve as a substrate for MPO in vitro7.
Type I interferons are involved in antiviral response and in bridging innate and adaptive immunity in normal individuals, but these type I interferons have been recognized as an important pathogenic factor in idiopathic SLE. They can be induced by viral particles or by DNA fragments, exposed namely after cell apoptosis or NETs15, and a chronic type I IFN production with a strong “type I IFN signature”, particularly in the skin, has emerged as a major marker in SLE and CLE16.
Reinforcing the role of type I interferons, there are reports of Di-LE in patients on treatment with IFN-α and IFN-α, but specially with IFN-α, estimated to occur in 0.15-0.7%17-19. These cases differ from Di-LE caused by other drugs as they lead to a higher frequency of anti-DNA antibodies (50%) and frequently have cutaneous involvement, also reinforcing the high involvement of type I IFN in CLE16.
Pathogenesis of Di-CLE
As for SLE, pathomechanisms involved in cutaneous disease are also multifactorial, but it is still uncertain whether similar pathways are responsible for cutaneous disease. The exception is the formation of NETs that are known to be involved in both conditions20, and very probably inducing type I interferon, whose expression in the skin is one of the highest among all organs involved in idiopathic LE15,16.
In some individuals, photosensitive drugs such as hydrochlorothiazide, terbinafine, and etanercept can trigger cutaneous LE in photoexposed areas7, particularly in patients who had already LE serological markers before drug exposure21. Apart from keratinocyte necrosis/apoptosis, photosensitive drugs increase Ro/SSA expression on the surface of keratinocytes, as in idiopathic subacute CLE, with consequent increased production of anti-Ro/SSA antibodies and cytotoxicity against these keratinocytes that express the Ro antigen on their surface22.
Chemotherapeutic agents may induce CLE through cell apoptosis, with release of nucleosides that will act as target for autoantigens and Type I -IFN production12.
Clinical manifestations of drug-induced LE
The time from starting the drug to the onset of lupus manifestations varies widely between drugs, but Di-LE usually occurs after months to years of exposure2. In the case of oncologic therapy, symptoms can occur within days of exposure7. The latency period may also be shorter (days or weeks) when the drug is reintroduced23.
Compared to the idiopathic SLE, Guicciardi et al. reported that patients with Di-SLE are considerably older and have more systemic manifestations, which are probably related to the advanced age and use of more medication24. Manifestations may affect many organs as in idiopathic SLE, but organ involvement is relatively specific to the offending drug8.
Constitutional symptoms such as fever, weight loss, anorexia, and fatigue1 and symptoms such as arthralgia/arthritis, myalgia, and serositis are the most frequent13. Cutaneous manifestations are less frequent, contrasting with 70% of skin involvement in idiopathic SLE25,26. An exception is cases induced by anti-TNFα drugs where the skin is involved in > 80% of cases2. Occasionally, sicca syndrome and Raynaud's phenomenon can also be found2.
Central nervous system, renal, gastrointestinal, and hematological manifestations are rare3 and also less frequent than in idiopathic SLE2, except for neurological involvement for quinidine (up to 30%)27 and lupus nephritis-like syndromes induced by hydralazine28-30, sulfasalazine, penicillamine, anti-TNFα, propylthiouracil, apixaban31,32, and also to phytotherapeutic agents33 and anti-neutrophil cytoplasmic antibodies (ANCAs) vasculitis associated with hydralazine30.
Di-CLE
Skin lesions in Di-CLE may present in an unspecified form or with the pattern of subacute or chronic CLE2. Subacute CLE is the most common, accounting for 70-80% of cases7 and occurs mostly in older women often associated with photosensitivity7.
Cutaneous features are very similar to the idiopathic subacute CLE24: erythematous, annular, papulosquamous lesions that do not usually evolve to scarring7 (Figs. 1 and 2), mainly on photoexposed areas, or occasionally in more protected areas34. An atypical and more widespread lesion distribution35, concomitant bullous and target lesions, vasculitis/purpura22, and erythema nodosum26 should raise the suspicion of a drug-induced case, as well as a change in the phenotype of the disease5.
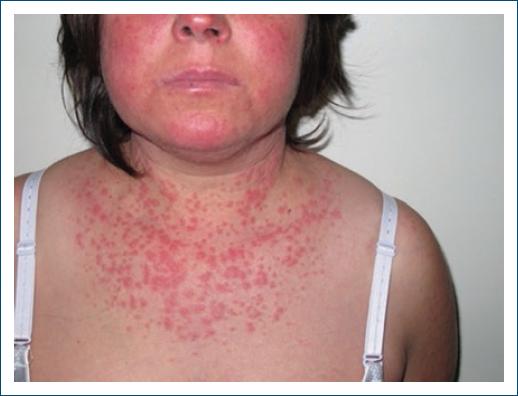
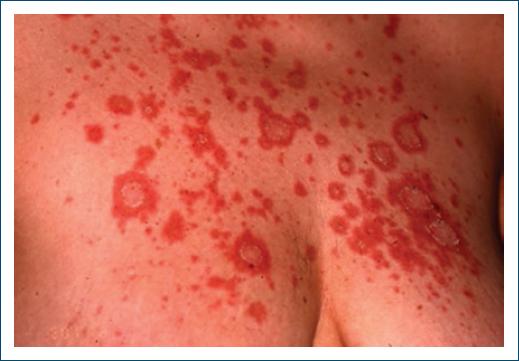
Figure 2 Subacute cutaneous lupus erythematosus induced by terbinafine in a patient with previous history of anti-Ro antibodies.
Chronic CLE is very rarely drug-induced7, corresponding to the least frequent subtype of Di-LE34. It occurs mostly in women, around the age of 40 years36, more often associated with 5-fluorouracil or anti-TNFα7, tends to have a slower onset (months to years) and resolves over months34. Lesions tend to occur more in photoexposed areas34 and are clinically similar to the idiopathic form7 (Fig. 3).
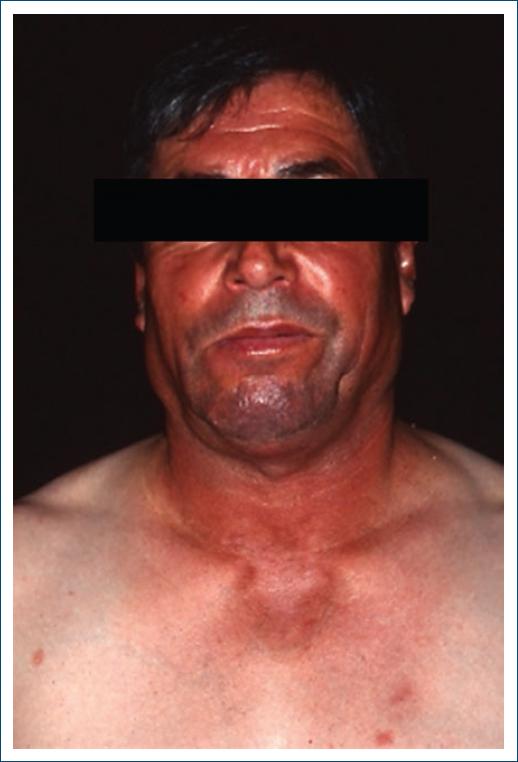
Drug-induced lupus tumidus and chilblain lupus have also been described2.
Histological characteristics
Differences between the histology of the idiopathic form of CLE and the drug-induced form have already been suggested, but studies are not concordant. Both forms are associated with focal vacuolization of the epidermal basal layer, perivascular and periadnexal lymphocytic infiltrates in the dermis, epidermal atrophy and edema, apoptotic keratinocytes and/or follicular obstruction22, and both can show granular IgM, IgG and C3 deposits at the dermoepidermal junction22.
Guicciardi et al. showed that subacute Di-CLE has no significant difference in the average number of eosinophils, basal layer cell liquefaction, keratinocyte necrosis, and depth and pattern of the inflammatory infiltrate but has less mucin deposition, more leukocytoclastic vasculitis, and IgM and C3 deposits in the basement membrane zone are less frequent24, but for Hillesheim et al., mucin deposition is similar in both forms37.
Laboratory findings
In Di-SLE erythrocyte, the sedimentation rate is high in up to 80% of patients8 whereas C-reactive protein tends to be normal, with the exception of quinidine-induced LE (high in 89% of cases)8. Anemia, leukopenia or thrombocytopenia are seldom found in Di-LE1,2, except for thrombocytopenia reported in 47% of quinidine-induced LE8 and pancytopenia frequently associated with hydralazine38. Coombs test is positive in < 30% and low complement levels are rare1, except in some quinidine-induced forms (low C3 and/or C4 in up to 1/3 of cases)2.
Autoantibodies to histone subunits, anti-histone antibodies (AHAs) are present in up to 95% of Di-SLE cases10 and are the hallmark and a very characteristic immunological marker of this form of Di-LE1, particularly anti-H2A-H2B antibodies in contrast to anti-H3 and H4 histone subunits more frequent in the idiopathic forms2. However, this differentiation is not commonly performed and also does not seem to add much diagnostic value39. AHAs can be IgG or IgM, although IgG is more prevalent in Di-LE38. Still, their pattern depends on the culprit drug, with procainamide associated with both IgG and IgM and hydralazine and chlorpromazine predominantly IgM38. Nevertheless, as AHAs are also present in > 50% of classic SLE, they cannot be used to distinguish drug-induced from idiopathic forms7. Furthermore, AHAs are not frequent in the cutaneous forms, and their presence is not synonymous with disease as they develop in 25% of patients treated with isoniazid, but only 1% of these develop clinical manifestations39.
Antinuclear antibodies (ANAs) are frequent2; however, negative ANAs should not exclude the diagnosis, especially if the patient has other LE-associated autoantibodies23. Anti-double-stranded deoxyribonucleic acid (anti-dsDNA) antibodies have been identified, mainly in those induced by anti-TNFα agents1. Anti-Smith (anti-Sm) antibodies are found in < 5% of Di-LE cases1, but have recently been described in six cases of Di-SLE1,32,40-43, one case of subacute Di-CLE44, and one of chronic Di-CLE36. Anti-phospholipid antibodies, such as lupus anticoagulant, have been found in a few cases13, but, in these cases, the drug may just be unmasking idiopathic SLE as in the case published by Sieiro Santos et al.45. Anti-cardiolipin antibody has also been reported in association with hydralazine, procainamide and minocycline8, metimazole10,32,45, apixaban10,32,45, and infliximab10,32,45.
ANCAs, both anti-proteinase-3 and anti-myeloperoxidase antibodies, have been identified in patients with minocycline and propylthiouracil-induced LE2, especially in patients with renal and pulmonary vasculitis14,30,46.
Anti-SSA/Ro (> 90%) and less frequently anti-SSB/La (< 50%) are similarly present in drug-induced and subacute CLE7, along with positive ANAs in 60-80% of cases47 by seldom AHAs48.
High autoantibody titers may persist for months to years after discontinuation of the offending drug, as opposed to clinical manifestations1.
Diagnosis of Di-LE
Although this entity has been gaining relevance over the years, there is still no consensus about diagnostic criteria7 and a temporal link with clinical, pathological, and serological findings compatible with LE contributes to establishing the diagnosis49. Borchers et al.8 have proposed the following criteria both for Di-SLE and Di-CLE: - continuous and sufficient exposure to a specific drug, - presence of at least one symptom consistent with LE, - no history of disease before starting treatment, - temporal relationship between the start of the drug and onset of the manifestations, and - discontinuation of the drug and the disappearance of the manifestations1. However, this definition still has some flaws, because there are reports of cases that persist despite drug discontinuation7 and cases of Di-LE in patients with a previous history of SLE5. Reappearance of drug reintroducing would contribute to the diagnosis, but this is not recommended39.
Diagnosing Di-LE can be more difficult for larger latency periods, simultaneous introduction of several drugs, new therapies with little information about their long-term effects4, and for treatment of neoplastic or autoimmune diseases, as these underlying conditions may be confounding factors49. In cases with several potentially suspected drugs, a probability algorithm such as Naranjo's can be used to guide which drug should be stopped first7.
Drugs frequently implicated in Di-LE
Over the years, an increasing number of drugs have been associated with Di-LE2, particularly in recent years both due to new therapies used in oncology and autoimmune diseases2 and due to new associations of “old” drugs7 (Table 1).
Table 1 List of drugs more commonly associated with cutaneous lupus erythematosus, divided by subacute, chronic, and other types of CLE
| Subacute cutaneous LE | Diuretics, hydrochlorothiazide7 |
| Diltiazem2, amlodipine35 | |
| ACE inhibitors4, beta-blockers7, phenytoin, carbamazepine lansoprazole, omeprazole, esomeprazole | |
| Anticholinergic agents: tiotropium7 | |
| Terbinafine, antiretroviral therapy4 | |
| Anti-TNFα, anti-IL17, anti-IL12/237 | |
| Anti-PD1 (nivolumab, pembrolizumab)/anti-PDL1 (atezolizumab), anti-CTLA4 (ipilimumab)7, | |
| Immunoglobulins G, leflunomide7 | |
| Mast cell inhibitors (mastinib), anti-CDK (palbociclib)51 | |
| Allopurinol, mitotane, pirfenidone4 Bupropion, ticlopidine, rosuvastatin, estroprogestatives7 | |
| Paclitaxel, tamoxifen51, doxorubicin, docetaxel, gencitabine7, taxanes, pemetrexed, hydroxyurea34, 5-FU compounds62, pazopanib, bevacizumab21 | |
| Topical treatments: terbinafine, imiquimod cream7, topical beta-blocker63 | |
| Chronic cutaneous LE | Fluorouracil compounds2, capecitabine, tegafur, and uracil/tegafur64 |
| Non-steroidal anti-inflammatory drugs7, | |
| Anti-TNFα (infliximab, etanercept, adalimumab, | |
| certolizumab pegol and golimumab7), Antifungals, intravenous immunoglobulin65 | |
| Other forms of cutaneous LE | Lupus tumidus: ustekinumab, bortezomide58 |
| “Chilblain lupus”: infliximab, adalimumab, etarnecept58 | |
| Rowell syndrome: terbinafine7 |
LE: lupus erythematosus.
Drugs can be classified as high or low risk1 or as high, medium, low, and very low risk of inducing SLE2 (Table 2) or into definitely (isoniazid, procainamide, and hydralazine), probably (phenytoin and carbamazepine), possibly (lithium and lamotrigine) and recently reported to induce Di-LE4,50. However, for instance, proton-pump inhibitors (PPIs) and terbinafine51 are not categorized.
Table 2 Drugs associated with systemic lupus erythematosus, with organization according to the risk of inducing the systemic type
| Very low risk | Statins, anti-TNFα2 |
| Minocycline4 | |
| Low risk | Carbamazepine, phenytoin, primidone, ethosuximide1 |
| Penicillamine, methyldopa, sulfasalazine, minocycline, isoniazid, chlorpromazine, propylthiouracil2 | |
| Captopril, acebutolol26 | |
| Intermediate risk | Quinidine2 |
| Isoniazid4 | |
| High risk | Procainamide (20-30%)4 |
| Hydralazine (5-10%)4 |
Anti-TNF α-induced LE
LE induced by tumor necrosis factor-alpha inhibitors (anti-TNF α) is rare (< 1%), it affects mostly women45,52, and in many cases, the drug just reveals a pre-existing LE2. It is considered distinct, as it sets up several exceptions to the typical features of Di-LE, but the three main forms of LE have been described36.
This seems to be a class effect but is particularly evident for infliximab45 and etarnecept53. These drugs induce: - apoptosis enhancing formation of autoantibodies against nuclear antigens8; - negative regulation on C-reactive protein and TNFα with consequent decrease in the expression of the adhesion molecule CD44 and reduced clearance of apoptotic material8; - and increase in type I interferon levels, which influences plasma cell differentiation51 – “cytokine shift” with suppression of T-helper 1 and increase of T-helper 2 cells, with B-cell activation and autoantibody formation45,54.
In TNFα-induced-LE cutaneous (Fig. 4), renal, cerebral, and hematological involvement is most commonly seen45,52 as well as other atypical manifestations such as hepatitis, pneumonitis, valvulitis, deep vein thrombosis, neuritis, and myositis have also been reported52.
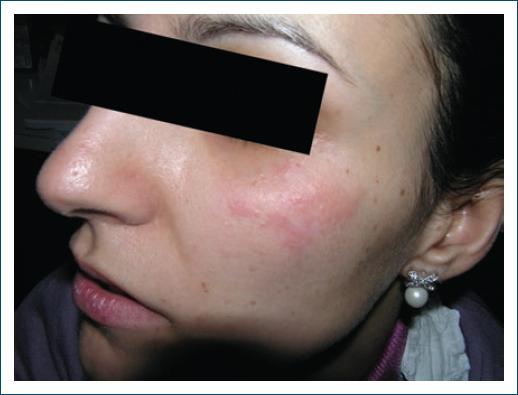
Figure 4 Drug-induced SLE: erythematous asymptomatic lesions on malar areas in a patient who developed ANA after use of anti-TNF-a for Crohn's disease.
ANAs and anti-dsDNA are very common (90% of cases)55 as well as anti-cardiolipin antibodies (25% of cases),45 but anti-histone antibodies are usually negative2. Anti-dsDNA antibodies are predominantly IgM with less systemic pathogenicity than the IgG antibodies found in idiopathic SLE, and therefore, this anti-TNFα-induced LE is less severe53. These autoantibodies are often present without clinically evident disease2. Hypocomplementemia is relatively frequent2.
In milder cases, patients can tolerate substitution to another anti-TNFα25,54, and some might tolerate treatment continuation, eventually adding immunosuppressive drugs45.
COVID-19 vaccine-induced LE
Given the need for rapid development of SARS-CoV-2 vaccines in response to the COVID-19 pandemic, there has not been sufficient time for studies about their adverse effects in the population and, recently, reports of vaccine-associated outbreaks of autoimmune diseases and vaccine-induced cases of LE have been reported42, as with previous vaccinations43,56. Nevertheless, these effects should not discredit vaccination56.
Patients with autoimmune diseases already have, ad initium, a higher propensity to develop complications from the vaccine. It has been reported that 1/3 of patients with idiopathic LE who received SARS-CoV-2 vaccination had an outbreak of their underlying autoimmune disease43. Still, in addition to disease exacerbations, Sagy et al. reported the case of three patients with SLE onset after vaccination with the Pfizer BNT162b2 mRNA vaccine, two of whom developed cutaneous manifestations43. Khanna et al. reported a case of SLE induced by the Pfizer BNT162b2 mRNA vaccine and reviewed eight other cases induced by the Pfizer, the Astra-Zeneca, and Moderna mRNA vaccines42. Most patients were female between 30 and 40 years old, and lupus involved the skin and the musculoskeletal system, followed by the renal and gastrointestinal systems42.
Possible mechanisms included molecular mimicry and the activation of toll-like receptors (TLRs) on antigen- presenting cells with production of specific autoantibodies43 or activation of TLRs by the viral mRNA, in conjunction with cytosolic inflammatory components, namely, the pyrin domain of the NLR family (NLRP3), leading to the onset of inflammation and autoimmunity56.
Reversibility and treatment of Di-LE
Clinical manifestations of Di-LE tend to resolve after drug discontinuation, the first recommended therapeutic step (2) that should be associated with lifestyle modifications such as smoking cessation and sun protection51.
If a lupus-like condition persists after drug withdrawal, treatment should be based on patient's manifestations2, but this treatment can often be reduced or even discontinued as symptoms resolve51.
In skin lesions, topical agents, such as corticosteroids or calcineurin inhibitors, are recommended2 or, when lesions are more generalized, systemic therapies such as anti-malarials, non-steroidal anti-inflammatory drugs (NSAIDs), and corticosteroids may be used2. Systemic immunosuppressants such as azathioprine, cyclophosphamide, mycophenolate mofetil2, or biologic therapeutics3 may be needed, particularly in Di-SLE with involvement of multiple organs and systems7.
Reintroduction of the culprit drug may be safe and effective in some cases with minor symptoms, but it is suggested that reintroduction should be associated with a short-term immunosuppressive treatment57. Maintenance of treatment with the causative drug has been described in patients under therapy with some anti-TNFα (infliximab and adalimumab), in most cases associated with systemic immunosuppressive agents58. This approach can be very useful, especially in patients with chemotherapy-induced LE34.
For some therapeutic classes, such as PPIs, thiazide diuretics, anti-TNFα, and chemotherapy agents, class effects have been reported59; thus, it may be necessary to contraindicate drugs of the same drug class7. There are also case reports of the disease recurrence after switching pharmacological classes, which reinforces the idea that there may be some genetic susceptibility for Di-LE60.
Resolution of clinical manifestations depends on several factors such as the type of drug, the type of clinical manifestations and their severity, and the characteristics of the patients, including their underlying disease2. Serological findings take longer resolution61, so they should not be used for therapy adjustment and evaluation2. When the manifestations are not reversible, it might mean that the condition originated from a pre-existing LE that was unmasked by a drug recently added to the patient's medication2.
Regular follow-up after resolution and in regards to the suspicion of a possible genetic susceptibility for the development of autoimmunity is recommended13.
Conclusion
Di-LE is indeed gaining importance in current clinical practice and, consequently, the number of studies published on this topic has been increasing, as well as the knowledge about the disease and main culprit drugs. However, most publications are isolated clinical case reports, with few studies adding new information on more precise diagnostic criteria and better knowledge of pathophysiology, characteristics of the implicated drugs, and their risk factors. This might allow, in the future, to screen patients more likely to develop the disease and avoid higher-risk drugs in more susceptible patients. In addition, a greater understanding of Di-LE may also contribute to explain pathomechanisms involved in the idiopathic forms and help develop more targeted treatments.

