










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med v.23 n.6 Porto nov. 2009
Câncer de Ovário Pequenas Células Tipo Hipercalcêmico (OSCCHT)
Relato de Um Caso
Nicolas S. Lazaretti*, Viviane Weiller Dallagasperina*, Luís Alberto Schlittler*, Rodrigo Ughini Villarroel*, Glenio Spinato*, Micheli Sandri Dettmer*, Rubens Rodriguez†
*Serviço de Oncologia do Hospital da Cidade de Passo Fundo, Rio Grande do Sul, Brasil; †Instituto de Patologia, Passo Fundo, Rio Grande do Sul, Brasil
Câncer de ovário pequenas células – tipo hipercalcêmico (OSCCHT) é um tumor raro, ocorre geralmente em pacientes entre 10 e 40 anos de idade, geralmente apresenta-se como tumor sólido unilateral, com hipercalcemia em 2/3 dos casos no momento do diagnóstico. Pelo seu padrão histopatológico similar a outras neoplasias, a realização de um painel de imuno-histoquímica detalhado é essencial para o diagnóstico.
Descreve-se um caso clínico de uma adolescente de 14 anos, queixando-se de aumento do volume abdominal, náuseas, vômitos, constipação, amenorréia e emagrecimento, com massa pélvica palpável indolor. Apresentava hipercalcemia e, na tomografia computadorizada, imagem sólido cística, volumosa na região hipogástrica e mesogástrica com impregnação heterogênea pelo contraste, a lesão foi ressecada com microscopia e estudo imuno-histoquímico compatível com OSCCHT. A paciente foi submetida a quimioterapia, com melhora importante inicialmente. Entretanto houve recidiva tumoral e evoluiu para óbito.
Palavras-chave: tumor de ovário; hipercalcemia; carcinoma de pequenas células.
Small-cell Carcinoma of the Ovary, Hypercalcemic Type
Small-cell carcinoma of the ovary, hypercalcemic type (OSCCHT) is a rare tumor that affects women between 10 and 40 years of age, generally presents as a unilateral solid tumor with two thirds of the cases are associated with hypercalcemia. at diagnosis. Its standard histopathological similar to other cancers, the holding a detailed panel immunohistochemical is essential to diagnosis.
We report a case in an 14-year-old teenager who presented with an abdominal tumor, nausea and vomiting, constipation, weight loss, amenorrhea, with a large abdominal tumor was palpated painless. Hypercalcemia and computed tomography showed a cystic solid image, bulky in hipogastric and mesogastric region with heterogeneous impregnation by contrast. The tumor was resected with microscopy and immunohistochemical study compatible with OSCCHT. The patient underwent chemotherapy, with signi?cant improvement initially. However there was tumor recurrence and she died.
Key-words: ovarian tumor; hypercalcemia; small-cell carcinoma.
INTRODUÇÃO
Câncer de ovário pequenas células – tipo hipercalcêmico (OSCCHT) foi descrito pela primeira vez em 1982 por Dickersin et al (1). É um tumor raro, que afeta mulheres entre 10 e 40 anos de idade (média de 23 anos) (2). Geralmente apresenta-se como tumor sólido unilateral, com hipercalcemia em 2/3 dos casos (2) no momento do diagnóstico (1,3,4). O quadro clínico constitui-se de: dor abdominal ou pélvica, distensão abdominal, polaciúria, fadiga, vômitos, dispareunia e alterações do hábito intestinal, mas alguns casos são assintomáticos. Apesar de haverem vários relatos na literatura de pacientes com sobrevida prolongada, a maioria das pacientes foram a óbito dentro de 1-2 anos após o diagnóstico (1,3,4,6).
A gênese do tumor e o mecanismo de desenvolvimento da hipercalcemia são desconhecidos. Young et al originalmente sugeriu que esse tumor teria uma origem epitelial (3,4). No entanto, outros autores relataram que ele não é originado de células germinativas, nem mesmo epiteliais (4,5).
O tratamento tem a cirurgia como pilar principal, necessitando freqüentemente de terapias adjuvantes devido ao padrão de disseminação. Há extensão peritoneal ao diagnóstico na maioria dos casos, com necessidade de terapia adjuvante mais extensa, nesta situação a radioterapia abdominal total pode ser preferível à radioterapia pélvica isolada (4). Existem evidências de benefício com o uso de quimioterapia à base de platina.
RELATO DO CASO
Paciente feminina, 14 anos, solteira, estudante, católica, natural e residente em Passo Fundo – RS. Foi avaliada na emergência de um hospital regional em Fevereiro de 2008, queixando-se de aumento do volume abdominal, náuseas, vômitos, constipação há 10 dias e amenorréia há 3 meses além de emagrecimento de 4 Kg em 15 dias. Negava febre, dor abdominal, alterações urinárias ou uso de medicações.
Ao exame físico geral encontrava-se hipocorada ++/4+, emagrecida, apresentava uma massa pélvica palpável no nível da sínfise púbica que estava indolor a palpação, fixa, com limites precisos. Realizado exame ginecológico que evidenciou ao toque vaginal: útero de volume normal, colo impérvio, anexo direito não palpável, anexo esquerdo endurecido e aumentado de volume. A paciente foi, então, internada e solicitado alguns exames com o objetivo de diagnosticar a patologia de base, os quais são descritos abaixo:
Laboratório: Hemograma: (Anemia normocítica e normocrômica) HB 10,5; HT 30,6; BHCG: < 2 UI/L;Amilase Pancreática 37 UI/L; Antigeno carcinoembriônico (CEA) 0,56 ng/ml; Alfa-fetoproteina (AFP) 4,0 ng/ml; Cálcio sérico: 16,1 mg/dl; Cálcio iônico: 2,1 mg/dl; Albumina: 4,0 g/dl; CA 125: 360,3 U/ml. Demais exames laboratoriais sem alterações.
Ultrassonografia de abdome: Massa heterogênea, hiperecóide com cerca de 14,6 x 13,0 x 8,5 cm em região hipogástrica na linha média. Estudo doppler evidenciou abundante vascularização com IR (Índice de Resistência vascular) 0,42. Presença de líquido livre na cavidade peritoneal.
Tomografia Computadorizada de Abdome: Massa heterogênea de 973 cm3, sólido cística, com contornos lobulados, ocupando a região hipogástrica e mesogástrica com impregnação heterogênea pelo contraste, determinando deslocamento cranial das alças intestinais além de compressão da porção terminal do ureter esquerdo, não há invasão de estruturas adjacentes. Esse exame é compatívelcom neoplasia ovariana. Presença delinfonodos aumentados de volume em espaço retroperitoneal, periaórticos e retrocavais. Líquido livre na cavidade (Figura 1).
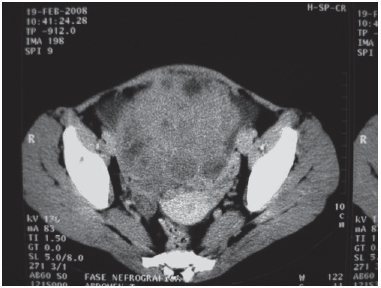
Fig. 1 - Tomografia computadorizada de abdome e pelve da paciente T. F. C. demonstrando grande massa em ovário esquerdo e múltiplas adenomegalias pélvica no pré-operatório.
Radiografia de tórax: Sem alterações relevantes.
Após exames foirealizadalaparotomia com ressecção do ovário esquerdo, tuba uterina esquerda e linfonodo ilíaco esquerdo. O líquido ascítico foi encaminhado para análise bioquímica e citopatológica. A paciente evoluiu sem intercorrência no pós-operatório imediato e, posteriormente, apresentou boa recuperação. O seu nívelsérico de cálcio iônico diminuiu para 1,32 mg/dl.
A análise da peça cirúrgica evidenciou neoplasia ma-ligna epitelióide de alto grau com mais de 20 mitoses por campo de grande aumento e áreas de necrose no ovário esquerdo e metástase para 1/1 linfonodo com extenção extracapsular. O estadiamento cirúrgico foi IIIC.
A análise microscópica e imunohistoquímica (Tabela 1) foi compatível com o diagnóstico de OSCCHT (Figura 2). A citologia do líquido ascítico foi positiva para células malignas.
Tabela 1 - Análise microscópica e imunohistoquímica.

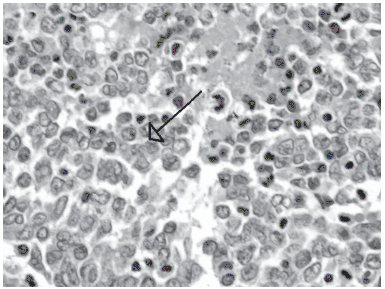
Fig. 2 -HE 400X Presença de massa de células redondas de citoplasma escasso, núcleos vesiculosos por vezes com pequenos nucléolos. Presença de foco de necrose tumoral na área indicada.
Após 2 ciclos de quimioterapia com etoposide associado a cisplatina, a paciente apresentou melhora clínica importante, recuperou 10% do peso, e a tomografia de abdome evidenciou diminuição de mais de 50% do volume das lesões retroperitoneais.
Neste momento foi realizada nova cirurgia com ressecção de útero, ovário direito, linfonodos da região ilíaca e obturadora, tuba uterina direita, omento maior, implantes peritoneais e análise do líquido ascítico. A análise das peças cirúrgicas demonstrou a presença de cisto teça-lutepinico em ovário direito, linfanedopatia reacional nos cinco linfonodos da região ilíaca e obturadora direita, câncer metastático em dois dos quatro linfonodos esquerdos, sem extenção extracapsular, implantes invasivos de carcinoma no peritôneo e exame citopatológico positivo para células neoplásicas malignas.
Três meses depois a paciente desenvolveu hidronefrose bilateral, o manejo incluiu cateterismo com colocação de cateter duplo J em ambos os ureteres e troca do esquema quimioterápico para gemcitabina associada a placitaxel.Devido a grande extenção tumorale a ausência de resposta clinica ao novo esquema de quimioterapia a paciente foi a óbito em Setembro de 2008, sete meses após o diagnóstico.
DISCUSSÃO
Durante a infância e a adolescência, os tumores de ovário são responsáveis por até 1% das neoplasias malignas. Entre essas, os carcinomas de ovário são extremamente raros.
Relatamos um caso de uma adolescente de 14 anos que se apresentou com um carcinoma de ovário tipo pequenas células subtipo hipercalcêmico. Essa doença geralmente é unilateral. Foi relatada pela primeira vez por Dickersin e colaboradores em 1982 em um relato de 11 casos associados à hipercalcemia. Essa doença é caracterizada por um curso rápido e letal independente da terapia agressiva, com uma taxa de sobrevida global de aproximadamente 10%. Em 1987 Ulbright e colaboradores demonstraram a histologia, imuno-histoquímica e sugeriram uma origem nas células germinativas (16).
Pelo seu padrão histopatológico similar a outras neoplasias, a realização de um painel de imuno-histoquímica detalhado se mostra de suma importância, onde sejam incluídos pelo menos os seguintes marcadores: Inibina, Antígeno de membrana epitelial (EMA) e Tumor de Wilms 1 (WTM1).
Patogenia
Macroscopicamente, o tumor é habitualmente sólido, tem diâmetro de 6 a 26 cm (média de 15 cm), com coloração cinza, áreas de hemorragia, necrose e degeneração cística. No caso relatado neste artigo, a massa tumoral possuía 14,6 cm no seu maior eixo, constituição sólido cística e hipervascularizada. Microscopicamente, as células tumorais mais comumente crescem num padrão difuso, são pequenas e redondas com escasso citoplasma, núcleos hipercromáticos e pequenos nucléolos (2). Espaços pseudo-foliculares que contenham substância eosinofílica estão presentes em 80% dos casos (2,3).
No painel de imuno-histoquimica, a inibina quase sempre é positiva, na grande maioria das pacientes com tumores do estroma gonadal e negativa em praticamente todas as pacientes com OSCCHT (5). S100, B72 (3), e desmina são geralmente negativos (2,3). Além disso, coexpressão de antígeno de membrana epitelial (EMA) e gene do tumor Wilms 1 (WT1) pode ter valor diagnóstico. Por último, a demonstração imuno-histoquimica do PTH ilustra a fisiopatologia da hipercalcemia, (13) embora algumas pacientes com imunorreatividade para PTH presente no tumor tenham cálcio sérico normal, e outras não tenham presença de PTH no tecido tumoral (8,9).
Estudos imuno-histoquimicos também mostram imunorreatividade para um ou mais marcadores epiteliais (pancitoqueratina, EMA, ou CAM 5.2) (2,3). Foiobservado imunorreatividade variável para vimentina, enolase, PTH e cromogranina.
Na análise microscópica do espécime cirúrgico desta paciente visualizou-se uma massa de células redondas de citoplasma escasso, núcleos vesiculosos, por vezes com pequenos nucléolos, presença de foco de necrose tumorale positividade para pancitoqueratina, confirmando o diagnóstico de OSCCHT.
Manifestações clínicas e diagnóstico diferencial
As manifestações clínicas da doença, em uma série de casos foram: dor (53%), edema ou distensão abdominal (41%), aumento da freqüência urinária (17,6%), fadiga (11,8%), vômitos (11,8%), dispareunia (11,8%) e mudanças no hábito intestinal (11,8%). Três pacientes (17,6%) eram assintomáticas (4).
O diagnóstico diferencial é amplo e inclui tumores de células da granulosa, tumor de células germinativas primitivas, linfoma, tumor neuroectodérmico primitivo, neuroblastoma, e metástases de carcinoma de pequenas células (tipo pulmonar), entre outros (2).
Tratamento
Cirurgia é o pilar principal da terapia, com quimioterapia e radioterapia frequentemente sendo utilizadas como adjuvantes. A melhor abordagem cirúrgica é desconhecida, mas, como a doença é unilateral em 99% dos casos, a ooforectoma unilateral é uma boa opção terapêutica apenas na doença localizada (3,4).
Tratamento adjuvante com quimioterapia à base de platina foi utilizado para a maioria das pacientes, com poucas recebendo radioterapia adjuvante. Éinteressante ressaltar que, uma paciente com doença volumosa, teve resposta completa com cisplatina e etoposide. Toxicidade foiconsiderávelcom regime de quimioterápicos conhecido com VCPBAE (associação de vimblastina, cisplatina, ciclofosfamida e etoposide) (4,10,11). Quimioterapia em altas doses com carboplatina e etoposide seguida de transplante autólogo de medula óssea também é uma alternativa de tratamento (15). O acúmulo da proteína p53 pode constituir uma possível explicação para a fraca resposta a tratamentos citostáticos habitualmente utilizados (13).
A pelve e o abdome foram os dois sítios mais comuns de recidiva. Dado o padrão de disseminação com citologia peritoneal positiva na maioria dos casos ao diagnóstico e os sítios de recidiva, a radioterapia abdominal total pode ser preferível à radioterapia pélvica isolada (4).
Prognóstico
O prognóstico é geralmente pobre. Na revisão de Young et al (3) 33% das pacientes com doença estádio IA estavam vivas e livres de tumor em média 5,7 anos após a cirurgia. Em contrapartida, apenas 10% dos pacientes com doença estádio IC e 6,5% das doentes com estádios II, III e IV estavam vivas e sem recorrência na última análise interina (3). Neste estudo, fatores prognósticos favoráveis para tumores na fase IA foram: idade maior de 30 anos, cálcio sérico pré-operatório normal, tumor inferior a 10cm, e ausência de componente de grandes células (3).
REFERÊNCIAS
1 -Dickersin GR, Kline IW, Scully RE. Small cell carcinoma of the ovary with hypercalcemia: a report of eleven cases. Cancer 1982;49:188-97.
2 -Chen F, Koenig C, Heller DS. A26-Year-Old Woman With Right Ovarian Mass. Arch Pathol Lab Med 2006;130:56-8.
3 -Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type: a clinicopathological analysis of 150 cases. Am J Surg Pathol 1994;18:1102-16.
4 -Harrison M.L, Hoskins P, Bois A, et al. Small cell of the ovary, hypercalcemic type—Analysis of combined experience and recommendation for management. A GCIG study Gynecologic Oncology 2006;100:233-8.
5 -McCluggage WG, Oliva E, Connolly LE, et al. An immunohistochemical analysis of ovarian small cell carcinoma of hypercalcemic type. Int J Gynecol Pathol 2004;23:330-6.
6 -Hamilton S, Beattie GJ, Williams AR. Small cell carcinoma of the ovary: a report of 3 cases and review of the literature. J Obstet Gynaecol 2004;2:169-72.
7 -Walt H, Hornung R, Fink D, et al. Hypercalcemic type of small cell carcinoma of the ovary: characterization of a new tumor line. Anticancer Res 2001;21:3253-9.
8 -Wynn D, Everett GD, Boothby RA. Small cell carcinoma of the ovary with hypercalcemia causes severe pancreatitis and altered mental status. Gynecologic Oncology 2004; 95:716-8.
9 -Matias-Guiu X, Prat J, Young RH, et al. Human parathyroid hormone-related protein (PTHrP) in ovarian small cell carcinomas. Cancer 1994;73:1878-81.
10 -Senekjian EK, Weiser PA, Talerman A, Herbst AL. Vinblastine, cisplatin, cyclophosphamide, and etoposide in the treatment of small cell carcinoma of the ovary. Cancer 1989;64:1183-7.
11-McCluggage WG. Ovarian neoplasms composed of small round cells: a review. Adv Anat Pathol 2004;11:288-96.
12 -Reed WC. Small cell carcinoma of the ovary with hypercalcemia: a report of a case of survival without recurrence 5 years after surgery and chemotherapy. Gynecol Oncol 1995;56:452-5. [ Links ]
13 -Peccatori F, Bonazzi C, Lucchini V, Bratina G, Mangioni Z. Primary ovarian small cell carcinoma: four more cases. Gynecol Oncol 1993;49:95-9.
14 -Distelmaier F, Calaminus G, Harms D, et al. Ovarian Small Cell Carcinoma of the Hypercalcemic Type in Children and Adolescents. Cancer 2006; 107:2298-306.
15 -Schleef J, Wagner A, Kleta R, et al. Small-cell carcinoma of the ovary of the hypercalcemic type in an 8-year-old girl. Pediatr Surg Int 1999;15:431-4.
16 -Ulbright TM, Roth LM, Stehman FB, Talerman.A, Senekjian EK. Poorly differentiated (small cell) carcinoma of the ovary in young women: evidence supporting a germ cell origin. Hum Pathol 1987;18:175-84.
Correspondência:
Dr.ª Viviane Weiller Dallagasperina
Universidade de Passo Fundo
Teixeira Soares, 817
CEP: 99010-080
Passo Fundo, RS, Brasil
e-mail: vividallagasperina@hotmail.com

