










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.25 no.1 Porto fev. 2011
ARTIGO DE REVISÃO
Hipertensão Pulmonar no Recém-nascido
Pulmonary Hypertension of the Newborn
Gustavo Rocha1
1Serviço de Neonatologia, Departamento de Pediatria, Hospital de São João EPE, Porto
RESUMO
A hipertensão pulmonar do recém-nascido é uma condição rara associada a várias situações clínicas e apenas tratada em centros especializados. Esta revisão descreve áreas conhecidas desta patologia, bem como algumas menos familiares. É abordada a fisiologia básica subjacente à hipertensão pulmonar e doença vascular. São descritas a avaliação diagnostica, as terapêuticas actuais ao nosso dispor bem como futuras terapêuticas.
ABSTRACT
Pulmonary hypertension of the newborn is a rare condition found in many clinical scenarios and only managed in specialized centers. This review describes the areas that are known in this condition and those that are less familiar. The basis physiology behind pulmonary hypertension and vascular disease are reviewed. The diagnostic evaluation as well as current and future therapies are described.
Introdução
A primeira descrição de hipertensão pulmonar no recém-nascido, por Gersony e colaboradores, em 1969, teve a designação de persistência de circulação fetal.1 Actualmente, uma vez que a placenta não está incluída na circulação neonatal, a designação hipertensão pulmonar persistente do recém-nascido (HPPRN) tem sido usada preferencialmente.2,3
A HPPRN ocorre quando a resistência vascular pulmonar não diminui imediatamente após o nascimento. Esta situação obriga à persistência de um shunt direito-esquerdo extra-pulmonar, através do foramen ovale e do canal arterial patente, impossibilitando a normal circulação pulmonar e enriquecimento do sangue em oxigénio, causando hipoxia arterial.3
A HPRN afecta principalmente o recém-nascido de termo ou quase termo, com uma incidência de aproximadamente 0,1 a 0,2% (1 a 2 em cada 1000 nados vivos).2,4 A prematuridade, incluído a inferior a 32 semanas, não exclui a possibilidade de HPPRN, uma vez que os factores desencadeantes podem existir na segunda metade da gestação.2,5 Não parece haver predomínio de raça ou género.2
São várias as condições que predispõem a HPPRN (Tabela 1).6 A Organização Mundial de Saúde, em 2003, efectuou uma revisão da nomenclatura e classificação da hipertensão pulmonar, que não sendo especificado recém-nascido, é apresentada na Tabela 2, como complemento desta revisão.7
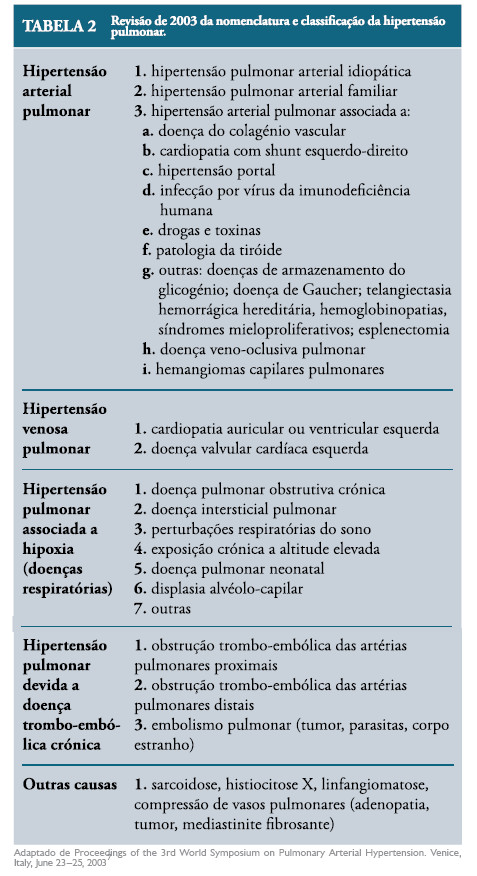
Alguns estudos demonstraram que a síndrome de aspiração de mecónio é a causa mais frequente de HPPRN, seguida da HPPRN primária.4,8 No entanto, nos últimos 10 anos tem-se verificado uma diminuição na frequência da síndrome de aspiração meconial, coincidente com a diminuição do número de gestações pós-termo.9 Por outro lado, a síndrome de dificuldade respiratória do recém-nascido pré-termo entre as 34 e as 37 semanas de gestação, após cesariana electiva, tem vindo a tornar-se uma causa frequente de HPPRN.3
Circulação fetal
No feto, a placenta assegura a oxigenação sanguínea. As duas artérias umbilicais levam sangue fetal à placenta, de onde o sangue regressa enriquecido em oxigénio pela veia umbilical, com uma PO2 de 30 a 40 mmHg. O fluxo da veia umbilical é orientado para o lobo esquerdo do fígado e para a veia cava inferior através do ductus venosus a nível do seio venoso portal. O sangue da veia cava inferior, antes da sua entrada na aurícula direita, resulta da junção do sangue da metade inferior do corpo, com o sangue drenado pelo ductus venosus e com o sangue vindo das veias hepáticas. A aurícula direita recebe também o sangue proveniente da veia cava superior. Devido a características anatómicas da aurícula direita, o sangue proveniente da veia cava inferior é orientado preferencialmente para a aurícula esquerda através do foramen ovale, juntando-se ao sangue venoso pulmonar. Este sangue, relativamente oxigenado, é de seguida, bombeado pelo ventrículo esquerdo para a metade superior do corpo, irrigando o coração e o cérebro.2 A maioria do sangue ejectado pelo ventrículo direito (90%) é desviada para a aorta descendente através do canal arterial (ductus arteriosus). Este shunt é condicionado pela existência de uma resistência vascular pulmonar elevada e uma resistência sistémica baixa (cerca de um décimo da resistência pulmonar, condicionada pela circulação placentária). Os restantes 10% do sangue do ventrículo direito são orientados para os pulmões através da artéria pulmonar. O circuito fica completo quando o sangue da aorta descendente retorna à placenta através das artérias umbilicais.2
Modificações respiratórias e hemodinâmicas na transição fetal-neonatal
Uma sequência de eventos ocorre imediatamente após o nascimento de modo a permitir uma adequada oxigenação no recém-nascido separado da placenta.
O pulmão fetal é um órgão cheio de líquido que não contribui para as trocas gasosas e oferece elevada resistência ao fluxo sanguíneo.10 A baixa tensão em oxigénio do fluxo sanguíneo pulmonar fetal e a libertação de vasoconstritores endógenos como a endotelina1 e o tromboxano, contribuem para a elevada resistência vascular pulmonar.11 Com o nascimento e início dos esforços respiratórios dá-se a expansão pulmonar, com saída de líquido alveolar e abertura do leito vascular. O oxigénio do ar inspirado tem efeito vasodilatador, resposta mais evidente após as 31semanas de gestação.12,13 A resistência vascular pulmonar sofre uma diminuição significativa, diminuindo em 50% a pressão na artéria pulmonar, associado a um aumento de 10vezesno fluxo sanguíneo pulmonar.14 O aumento do fluxo sanguíneo pulmonar facilita as trocas gasosas. O oxigénio é o estímulo mais importante para a vasodilatação do leito vascular pulmonar, mas também a diminuição da PaCO2 e aumento do pH contribuem para esta resposta.13 Estes estímulos fisiológicos promovem a libertação de vários vasodilatadores, incluindo mediadores derivados do endotélio, óxido nítrico e prostaglandinas.15-18
A enzimas intetase do óxido nítrico endotelial (eNOs) tem um papel crítico na transição da circulação pulmonar, através da produção de óxido nítrico. Esta enzima, na presença de oxigénio, converte l-arginina em l-citrulina e óxido nítrico. O oxigénio estimula a libertação de óxido nítrico directamente18 e indirectamente através do aumento da oxidação fosforilativa e libertação de ATP (adenosina trifosfato) de eritrócitos fetais oxigenados.18,20 O aumento e maturação da enzima eNOs ocorrem e são críticos no final da gestação, uma vez que o óxido nítrico não é armazenado e a sua síntese requer elevada expressão enzimática.21 O óxido nítrico inicia rápida vasodilatação por estimulação da enzima solúvel, guanilate ciclase, dentro dacélulado músculo lisovascular,a qual, por sua vez, converte moléculas de guanosina trifosfato nucleótido em guanosina monofosfato cíclico (GMPc). O aumento intra-celular de GMPc diminui o influxo de cálcio e produz relaxamento do músculo lisovascular. A fosfodiesterase tipo 5, no interior da célula muscular lisa vascular, degrada o GMPc e limita a duração da vasodilatação. O óxido nítrico tem, também, pa-pel promotor no crescimento vascular pulmonar in utero, em resposta ao factor de crescimento endotelial vascular (VEGF).22 Este crescimento vascular é acompanhado por crescimento alveolar.23 Os peptídeos natriuréticos, ANP (peptídeo natriurético auricular) e peptídeo natriurético tipo B (BNP) aumentam os níveis de GMPc intra celular no músculo lisovascular, contribuindo para a vasodilatação arterial pulmonar.24 De qualquer modo, o seu papel na transição fetal neonatal não está, ainda, completamente esclarecido. O sistema peptídeo natriurético –GMPc parece funcionar em paralelo com o sistema óxido nítrico-GMPc. O sistema das prostaglandinas também é activado, ao nascimento, no pulmão fetal.25 Este sistema é mediador de vasodilatação vascular pulmonar, complementando o sistema do óxido nítrico. A prostaciclina (PGI2) é o mais potente dos vasodilatadores do grupo das prostaglandinas e activa a enzima adenilate ciclase na célula muscular lisa vascular, que converte ATP em AMPc (adenosina monofosfato cíclica), que diminui o influxo de cálcio intracelular e consequente relaxamento vascular. A enzima fosfodiesterase tipo 3 (PDE-3) degrada o AMPc e limita a duração da vaso dilatação induzida por este mediador.
Biologia vascular alterada na HPPRN
A HPPRN pode resultar de insuficiente desenvolvimento pulmonar incluindo a sua árvore vascular (hérniadia fragmática congénita, hipoplasia pulmonar), má adaptação do leito vascular na transição fetal-neonatal (várias condições, como stress perinatal, hemorragia, hipoxia, asfixia, aspiração, hipoglicemia) ou desenvolvimento anómalo do leito vascular pulmonar de causa conhecida ou desconhecida. Contudo, mesmo quando há evidência de um evento perinatal (ex: aspiração de mecónio) a causa da HPPRN é um processo intrauterino de alguma duração.3
A falência da vasodilatação vascular pulmonar após o nascimento pode resultar de inadequada oxigenação ou expansão pulmonar, bem como da falência na libertação de óxido nítrico e/ ou prostaglandinas. Alterações bioquímicas no sistema óxido nítrico – GMPc incluem diminuição da expressão de eNOs,26 diminuição da disponibilidade de arginina,27 e diminuição da produção de óxido nítrico avaliada pela diminuição da excreção dos metabolitos urinários.27 O modelo animal de HPP mostra uma consistente diminuição na expressão de eNOs nas artérias pulmonares.28,29 Este facto pode ser devido a alteração numa ou mais etapas do sistema óxido nítrico–GMPc. É o caso da inibição competitiva do análogo natural da arginina, arginina dimetil assimétrica, que competindo com a arginina leva a diminuição da produção de óxido nítrico.30,31 Também, o aumento dos níveis do peptídeo endotelina1 inibea enzima eNOs32,33 e o radical livre superóxido presente nas células vasculares pode inactivar a acçãodo óxido nítrico causando hipertensão pulmonar,34,35 bem como a diminuição da expressão de gualinate ciclase, levando a diminuição da disponibilidade de GMPc.36 A exposição pré-natal a anti-inflamatórios não esteróides diminui a acção da cicloxigenase eleva a diminuição da síntese de prostaglandinas vasodilatadoras aumenatando o risco de HPPRN.37,38 O uso de anti-inflamatórios não esteróides leva a constrição e/ou encerramento do canal arterial fetal, elevação das pressões nas artérias pulmonares, persistência de shunts direito-esquerdo após o nascimento, diminuição da libertação de óxido nítrico pelas artérias pulmonares e aumento de radicais livres de oxigénio como superóxido, peróxido de hidrogénio e peroxinitrito.28,29,34,35,39-41 Também se verificou que a hipertensão pulmonar intra-uterina por constrição do canal arterial leva a alterações no desenvolvimento da vascularização pulmonar e alveolar.42
Recentemente, um estudo epidemiológico sugeriu uma incidência aumentada de HPPRN na exposição pré-natal aos anti-depressivos inibidores da recaptação selectiva de serotonina, durante o terceiro trimestre de gestação.43 Contudo, esta associação não foi confirmada por outro estudo,44 pelo que mais estudos são necessários para definir o efeito dos inibidores da recaptação selectiva da serotoninana circulação pulmonar fetal.
Tanto para os anti-inflamatórios não esteróides como para os inibidores da recaptação selectiva de serotonina, alguns recém-nascidos com exposição pré-natal não desenvolveram HPPRN, sugerindo uma susceptibilidade biológica com papel na patogénese dadoença.3
Diagnóstico de HPPRN
No recém-nascido de termo ou quase termo, hipoxémia significativa e lábil, desproporcional à patologia pulmonar, quando existe, é sugestivo, mas não diagnóstico de HPPRN.6 Dependendo da causa, o recém-nascido pode demonstrar-se gravemente doente após o nascimento, ou por outro lado, a instalação dos sinais clínicos pode ser insidiosa. Estes incluem sinais de dificuldade respiratória com gemido, adejo nasal, retração esternal, taquipneia, taquicardia, cianose e choque. Uma auscultação cardíaca anormal revelando um sopro sistólico de regurgitação tricúspide e um S2 proeminente pode estar presente, contudo não é diagnostica de HPPRN.3 Muitos dos sinais clínicos de hipertensão pulmonar podem estar presentes em doentes com cardiopatia congénita cianótica, pelo que uma abordagem sistemática do doente hipoxémico é fundamental. É importante reconhecer que a utilização de altas concentrações de oxigénio e vasodilatadores pulmonares podem agravar a perfusão sistémica num recém-nascido com cardiopatia esquerda ductus dependente como estenose aórtica crítica, interrupção do arco aórtico, coartação da aorta ou coração esquerdo hipoplásico.3
O estudo inicial inclui uma radiografia de tórax e análise dos gases do sangue arterial. Contudo, uma radiografia de tórax revelando patologia parenquimatosa pulmonar, não exclui uma cardiopatia congénita.3
Dependendo do grau e localização do shunt direito esquerdo, vários graus de oxigenação podem ser observados em locais pré-ductais (artérias radial e temporal direitas) e pós-ductais (artéria umbilical ou de uma extremidade inferior). Uma saturação pré-ductal 20 mmHg superior é, habitualmente, considerada significativa (ou 5% superior por oximetria de pulso). No entanto, a ausência deste gradiente não exclui HPPRN, uma vez que o shunt predominantemente auricular não cria esta diferença. Gradientes detectáveis ocorrem quando o shunt direito-esquerdo é ao nível do canal arterial.2 A diferença nas saturações pré e pós ductais de oxigénio podem, também, estar associadas a coartação da aorta.
O ecocardiograma é fundamental para a correcta avaliação do recém-nascido com hipertensão pulmonar. O cateterismo cardíaco está reservado para casos seleccionados cuja investigação anterior não permita o diagnóstico.2 Na prática clínica o diagnóstico e seguimento do doente com hipertensão pulmonar baseiam-se nos dados ecocardiográficos. A avaliação diagnóstica na HPPRN encontra-se resumida na Tabela 3.45 A associação entre hipertiróidismo neonatal e hipertensão pulmonar já foi descrita na literatura,46-48 pelo que a avaliação da função tiróideia está indicada quando não existe uma causa evidente como síndrome de aspiração ou pneumonia congénita. Um caso fatal de hipotiróidismo congénito associado a hipertensão pulmonar em que a doente apresentava uma mutação (1207F) no gene do factor 1 de transcrição tiróideia (TITF1/NKX2.1) (cromossoma 14q13), foi descrito por Maquet E e colaboradores. Estes autores propõem que sempre que a TSH (thyroid stimulating hormone) se encontre elevada em associação com insuficiência respiratória não explicada deve ser efectuada a sequenciação do gene TITF1/NKX2.1.49
Também, a associação de hipertensão pulmonar com doenças auto-imunes e do tecido conjuntivo, nomeadamente a esclerodermia, tem sido descrita após o período neonatal.50 No entanto, a possibilidade de mecanismos autoimunes como causa de hipertensão pulmonar neonatal já foi colocada por Morse JH e colaboradores.51
A associação entre infecção pelo vírus da imunodeficiência humana e hipertensão pulmonar é rara, mas bem documentada no adulto,52 no entanto, descrições na criança pequena começam a surgir na literatura.53
Hipertensão pulmonar familiar
A hipertensão arterial pulmonar familiar representa menos de 6% dos casos idiopáticos.54 A doença é autossómica dominante com penetração incompleta e, em 50% dos casos, ligada a mutações no gene do receptor tipo II de proteinas morfogenéticas do osso (BMPR2), encontradas no cromossoma 2q31-32.55-59 As proteínas morfogenéticas do osso são parte da superfamília de citocinas TGF ß (transforming growth factor ß). Mutações no BMPR2 conferem 15% a 20% de probabilidade de desenvolver hipertensão pulmonar arterial durante a vida. A forma familiar da doença aparece mais cedo nas gerações sucessivas (antecipação genética) e é mais comum no sexo feminino.
Mutações no gene do receptor ALK-1 (activin receptor-like kinase) foram encontradas em doentes com hipertensão pulmonar associada a telangiectasia hemorrágica hereditária.60 Provavelmente outros genes permanecem por ser identificados e, uma vez que apenas 15% a 20% dos indivíduos afectados de mutação BMPR2 apresentam hipertensão pulmonar, outros mecanismos estão envolvidos na génese da doença.
Tratamento
A prevenção, sempre que possível, permite minorar os danos da HPPRN, sendo exemplos a identificação atempada de situações de risco, como sinais de sofrimento fetal ou de líquido amniótico com mecónio, bem como a adequada reanimação no bloco de partos de um recém-nascido com asfixia.
O tratamento da HPPRN deve ser efectuado em centros especializados por equipa multidisciplinar experiente.45 A resposta ao tratamento é menos previsível na criança, pelo que a monitorização contínua e possíveis rápidas modificações nas atitudes terapêuticas podem ser necessárias.45 A investigação da causa subjacente e o tratamento direccionado devem ser iniciados o mais precocemente possível.61
O tratamento tem como objectivo prevenir lesões orgânicas causadas pela hipoxia e barotrauma.6 Em simultâneo com o tratamento da doença de base devem ser corrigidos todos os desequilíbrios associados como hipovolémia, hipo ou hipertermia, hipoglicemia, acidose, policitemia, anemia, pneumotórax, hipotensão sistémica, hipocalcémia e hipomagnesemia. É importante manter uma adequada resistência vascular sistémica, enquanto se tenta selectivamente diminuir a resistência vascular pulmonar.6 É importante manter uma normovolémia, pois a hipovolémia agrava o shunt direito-esquerdo.6
Manipulação mínima e sedação
Devido à lábil estabilidade clínica e de oxigenação, pequenos estímulos podem provocar deterioração clínica importante. A manipulação deve ser mínima, as aspirações endotraqueais não devem ser efectuadas por rotina mas apenas se indicadas. Todos os estímulos, nomeadamente luminosos, auditivos e tácteis devem ser reduzidos ao mínimo.
O uso de sedativos e relaxantes musculares tem sido prática generalizada, com o objectivo de minimizar variações na oxigenação e facilitar a ventilação. No entanto, esta prática não foi ainda testada em estudos aleatorizados.3 Estes fármacos apresentam efeitos ad-versos e frequentemente induzem hipotensão, edema generalizado e deterioração da função pulmonar com o uso prolongado. A hipotensão é mais comum quando são combinados sedativos com relaxantes musculares. Os relaxantes musculares têm sido associados a maior incidência de défice auditivo nos sobreviventes de HPPRN, contudo o mecanismo desta associação é desconhecido.62 Os diuréticos, frequentemente utilizados no tratamento do edema secundário ao relaxamento muscular, também se associam ao risco de défice auditivo.63 Embora a sedação possa ser necessária para o conforto do doente ventilado, alguns auto-res não recomendam o uso de relaxantes musculares por rotina mas aceitam o seu uso por necessidade por períodos não superiores a 48 horas.3
Ventilação mecânica
A ventilação mecânica facilita o recrutamento alveolar e a expansão pulmonar, melhorando a relação ventilação/perfusão. A estratégia ventilatória deve recrutar as áreas atelectásicas evitando simultaneamente a sobre distensão. Manter a PaO2 entre 60 e 90 mmHg é importante para a adaptação pós-natal e não há evidência de que uma PaO2 superior a 100 mmHg cause mais redução na resistência vascular pulmonar.3 Inicialmente é sensato usar uma fracção de oxigénio no ar inspirado (FiO2) de 100%, e efectuar uma diminuição gradual e lenta. O objectivo é manter uma adequada e estável oxigenação com o mínimo de pressões. Este objectivo consegue-se habitualmente com a ventilação convencional. Quando o recém nascido não consegue ser adequadamente oxigenado com a ventilação convencional, a ventilação por alta frequência oscilatória deve ser considerada precocemente.
A hiperventilação deve ser evitada e a pressão parcial arterial de dióxido de carbono (PaCO2) deve ser superior a 30 mmHg, e valores de 40-50 mmHg, ou mesmo superiores, são aceitáveis desde que a oxigenação seja adequada.64
A prática tradicional de manter uma PaO2 elevada (>100mmHg) e uma PaCO2 baixa, de modo a provocar vasodilatação pulmonar não tem demonstrado benefício e pode ser potencialmente lesiva para a perfusão pulmonar e cerebral.3 O efeito da hiperventilação na circulação pulmonar parece estar relacionada com a elevação do pH.65 A hipocarbia e a alcalose diminuem a perfusão cerebral e associaram-se a défice auditivo e lesões neurológicas.66,67
A hiperventilação e a alcalose induzem hipocalcémia, disfunção miocárdica e hipotensão sistémica. A alcalose diminui a libertação de oxigénio pela hemoglobina, o que pode potencialmente diminuir a entrega de oxigénio aos tecidos.3
Surfactante
A utilização de surfactante exógeno facilita a expansão alveolar na doença parenquimatosa. O efeito benéfico do surfactante tem sido documentado em casos de síndrome de aspiração meconial, sépsis com pneumonia e doença das membranas hialinas.64,68
Agentes vasopressores e cardiotónicos
O suporte vasopressor deve ser considerado um componente essencial na abordagem da HPPRN. Uma pressão sistémica adequada (> 40 mmHg) é necessária para diminuir o shunt direito-esquerdo, manter um adequado enchimento do ventrículo direito e um débito cardíaco adequado.3 A dopamina é o agente mais utilizado neste propósito. A dobutamina, em-bora melhore o débito cardíaco, tem menor efeito vasopressor. A milrinona, um inibidor da fosfodiesterase tipo 3, tem sido utilizada algumas vezes no tratamento da hipotensão e melhoria do débito cardíaco.64 Recentemente foi demonstrado que o uso de noradrenalina se associou a aumento da pressão sistémica e melhoria da oxigenação na HPPRN.69 O hematócrito deve ser mantido acima dos 35%.70
Terapêutica com óxido nítrico inalado
O óxido nítrico inalado (NOi) foi um marco na terapêutica vasodilatadora pulmonar.3 Estudos clínicos controlados demonstraram que o NOi melhora a oxigenação de uma proporção significativa de recém nascidos de termo e quase termo (> 34 semanas de gestação) com HPPRN. O seu uso está aprovado, desde o ano 2000, pela US Food and Drug Administration. O NOi atinge o espaço alveolar e difunde para o músculo liso da parede das artérias pulmonares adjacentes. O NOi causa vasodilatação através do aumento do GMPc intracelular no músculo liso. Uma vez que é preferencialmente distribuído aos segmentos ventilados (e não aos atelectásicos), provoca uma maior perfusão destes segmentos, pelo que um bom recrutamento alveolar é importante na HPPRN. À medida que o NO progride através da parede vascular até ao lúmen arterial, é inactivado pela hemoglobina, ficando o seu efeito limitado à circulação pulmonar.3
A melhoria na oxigenação é geralmente evidente em minutos após iniciar a medicação, o que facilita a oxigenação do doente severamente hipóxico. No en-tanto, já tivemos casos cuja resposta se verificou entre as quatro e as cinco horas após o inicio da terapêutica. Vários estudos demonstraram que o NOi diminui a necessidade de ECMO (extracorporeal membrane oxigenation) e mortalidade no recém-nascido de termo e quase termo com HPPRN e insuficiência respiratória hipóxica.72-76 O NOi melhora a oxigenação em cerca de 70% ou mais dos recém-nascidos com HPPRN, sendo as melhores respostas observadas nas formas idiopáticas.8,72,75
Estudos prévios sugerem a dose inicial de 20 ppm como a mais adequada, com efeito verificado entre os cinco e os 20 ppm.8 Doses superiores a 20 ppm não demonstraram mais eficácia e associaram-se a mais efeitos adversos.72, 77 No nosso serviço, alguns casos responderam a doses tão elevadas como 40 ppm. O inicio do NOi deve ser antes ou logo no início da insuficiência respiratória, quando se obtêm dois índices de oxigenação igual ou superior a 20.3 Existem três efeitos adversos potenciais do NOi: (1) metemoglobinemia, gerada pela oxidação da hemoglobina pelo NO (manter < 2%); (2) exposição ao dióxido de nitrogénio gerado pela reacção do NO com o oxigénio (manter < 0,5 ppm); (3) inibição da agregação plaquetária pelo NO. Estes efeitos adversos surgem habitualmente com doses de NOi superiores a 40ppm.3 A descontinuação da terapêutica com NOi pode provocar vasoconstricção rebound. A descontinuação só deve ser iniciada quando a FiO2 é de 0,60, e deve ser gradual e lenta (1 ppm ou menos, sobretudo a partir dos 5 ppm).3,70 A duração da terapêutica varia com a etiologia, mas geralmente inferior a 5dias.70 A dependência prolongada de NOi está associada a anomalias pulmonares subjacentes, como hipoplasia pulmonar ou displasia alvéolo-capilar. Apesar da terapêutica com NOi ser eficaz na HPPRN, deve ser considerada como uma estratégia clínica global. A falência da terapêutica com NOi ou a não retirada do NOi em cinco dias obriga a uma revisão clínica do doente bem como de toda a estratégia terapêutica. Recomenda-se a utilização de NOi antes da exposição prolongada a elevadas fracções de oxigénio inspirado ou suporte ventilatório máximo. A exposição a oxigénio a 100%, mesmo que por breves períodos pode induzir disfunção vascular, aumentando o stress oxidativo e impedindo a resposta subsequente ao NOi.78 O NOi permite uma rápida diminuição da FiO2 e diminui o stress oxidativo no modelo animal de HPPRN.79 A terapêutica com NOi não tem sido eficaz em muitos casos de hérnia diafragmática congénita, apesar de evidência clínica e ecocardiográfica de HPPRN.80 Também não diminuiu a mortalidade ou a necessidade de ECMO (extracorporeal membrane oxygenation) em recém-nascidos com hérnia diafragmática congénita.80 Por outro lado, a terapêutica com NOi não demonstrou eficácia em cerca de 30% dos casos de HPPRN.8,72
Sildenafil
A fosfodiesterase 5 (PDE5) é abundante no tecido pulmonar e degrada o GMPc (este causa captação de cálcio para o retículo sarcoplasmático que é usado na vasodilatação do músculo arterial pulmonar). O sildenafil, um inibidor da PDE5, prolonga a semi-vida do GMPc e teoricamente potencia a acção do óxido nítrico endógeno e inalado.3 Estudos não controlados demonstraram que o sildenafil tem efeito sinérgico como NOi. Alguns casos clínicos demonstraram que o sildenafil atenua a hipertensão pulmonar rebound após retirada do NOi em doentes com cardiopatia congénita.81
Um pequeno estudo aleatorizado com sildenafil oral versus placebo foi interrompido após a morte de cinco entre seis recém-nascidos do grupo placebo comparado com a morte de um em sete no grupo de sildenafil.82 Neste estudo verificou-se melhoria na oxigenação no grupo com sildenafil, entre as seis e as 12 horas após a primeira dose. Não foi documentada hipotensão arterial neste estudo. Tem também sido questionada a possibilidade de maior risco de retinopatia da prematuridade. O sildenafil endovenoso, não disponível para uso clínico, tem sido associado a hipotensão sistémica.83
Estudos experimentais no animal demonstraram que o verdenafil, outro inibidor da fosfodiesterase, tem mais eficácia que o sildenafil in vitro. 84,85
De qualquer modo, a experiência no recém-nascido é ainda restrita sendo necessários estudos mais alargados de modo a poder avaliar os riscos e os benefícios.
Agonistas da prostaciclina
A prostaciclina (PGI2) é um vasodilatador endógeno com acção no músculo liso via AMPc, causando vasodilatação pulmonar e sistémica.61 Tem um efeito sinérgico com o NOi, uma vez que actua por outro mecanismo, podendo ser utilizada em simultâneo, ou na retirada do NOi.61 Os análogos da prostaciclína podem ser utilizados por via endovenosa (epoprostenol, treprostinil), por via inalatória (iloprost) ou oral (beraprost). A maior experiência é com o epoprostenol, que requer perfusão contínua devido à sua curta semi-vida (< 6 minutos) e na criança e adulto associou-se a vários efeitos secundários indesejáveis, incluindo cefaleias, dores, eritema cutâneo, trombocitopenia e hipotensão sistémica.86 A interrupção da perfusão pode causar rápido aumento da resistência vascular pulmonar, colapso hemodinâmico e morte.86
A prostaciclina inalada tem uma vasodilatação pulmonar mais selectiva e requer administração contínua devido à curta semi-vida. No entanto, o agonista iloprost apresenta maior semi-vida e pode ser administrado por nebulização intermitente (cada duas horas). Embora não existam estudos da sua eficácia no recém-nascido, o seu efeito parece similar ao da prostaciclina inalada.3 A prostaciclina inalada já foi usada com êxito no recém-nascido com HPPRN refractária ao NOi.87
Não existe experiência com o beraprost no recém-nascido, no entanto a sua eficácia parece menor que a dos análogos endovenosos.61
Milrinona
O inibidor da fosfodiesterase 3 (PDE3), milrinona, demonstrou melhoria da hipertensão pulmonar e função cardíaca no pós-operatório de cardiopatia congénita.88 A perfusão de milrinona tem sido testada em estudos não controlados. Bassler e colaboradores89 e McNamara e colaboradores90 demonstraram uma elevação na PaO2 e diminuição do índice de oxigenação em resposta à perfusão de milrinona em recém nascidos com hipertensão pulmonar refractária a NOi. Se esta melhoria foi devida à acção da milrinona ou à resolução espontânea da doença só poderá ser avaliada em estudos aleatorizados.
O efeito da milrinona no AMPc pode ser aditivo ao da prostaciclina inalada e complementar ao NOi.3
Antagonistas dos receptores da endotelina
A endotelina 1 (ET-1) actua nos receptores ETA e ETB das células musculares lisas arteriais pulmonares.61 A activação de ETA e ETB no músculo liso causa vasoconstrição e a activação da ETB no endotélio liberta NO causando vasodilatação. Este balanço entre vasodilatação e vasoconstrição parece estar alterado em doente com HPPRN.61
O bosentan é um antagonista não selectivo dos receptores da endotelina 1 (ETA e ETB) que demonstrou melhoria hemodinâmica e da qualidade de vida em adultos e crianças com hipertensão pulmonar.61 Cerca de 10% dos doentes apresentaram toxicidade hepática. A experiência com o uso de bosentan no recém-nascido é escassa.91,92
O sitaxsentan, um antagonista selectivo ETA foiusado com sucesso no adulto com hipertensão pulmonar, sem evidência de toxicidade hepática. Não existe experiência actual no recém-nascido.61
Oxigenação por membrana extracorporal
A oxigenação por membrana extracorporal (ECMO) tem indicação no recém-nascido acima das 34 semanas de gestação com HPPRN sem resposta a outras terapêuticas e que reúne critérios para ECMO. O recurso à ECMO tem vindo a diminuir com a maior utilização do NOi, novos fármacos disponíveis para tratamento da HPPRN, optimização da ventilação mecânica incluindo ventilação com alta frequência, uso de surfactante exógeno e todo o suporte nutricional e metabólico. Ultimamente parece ter sido mais utilizada em casos de HPPRN por hipoplasia pulmonar secundária a hérnia diafragmática congénita.3
Terapêuticas futuras
Estudos recentes no modelo animal demonstraram que o stress oxidativo contribui para a falta de vasodilatação arterial pulmonar e resposta diminuída ao NO. O radical livre superóxido é um vasoconstritor e reage com o NO impedindo a vasodilatação. A utilização de superoxido dismutase recombinante diminuiu a vasoconstrição pulmonar e melhorou a resposta ao NO endógeno no modelo animal de HPPRN.79
A administração antenatal de betametasona reduz o stress oxidativo e melhora a resposta ao NO no modelo animal de HPPRN.93 A utilização de betametasona antenatal entre as 34 e as 37 semanas de gestação encontra-se actualmente em estudo pelas unidades da National Institutes of Child Health and HumanDevelopment (NICHD).3
A L-arginina é um substrato para a síntese de NO endógeno e a sua suplementação encontra-se em estudo como alternativa à terapêutica com NOi na HPPRN.61
Hipertensão pulmonar no recém-nascido pré-termo
A HPPRN tem sido documentada por ecocardiografia no recém-nascido pré-termo com idade gestacional inferior a 30 semanas com hipoxémia refractária ao tratamento com surfactante exógeno.5 Os factores de risco identificados foram a ruptura prolongada de membranas, a hipoplasia pulmonar e a restrição de crescimento intra-uterino.
A hipertensão pulmonar tem vindo, também, a ganhar crescente reconhecimento na doença pulmonar crónica da prematuridade (displasia broncopulmonar) em sobreviventes de prematuridade extrema.94 A diminuição do número de vasos sanguíneos pulmonares, a arquitectura pulmonar alterada, episódios de hipoxémia e hipercápnia contribuem para o desenvolvimento da hipertensão pulmonar na doença pulmonar crónica do pré-termo. Um estudo retrospectivo observacional de 42 recém-nascidos prétermo com displasia broncopulmonar demonstrou severa hipertensão pulmonar em 43%, diagnosticada a uma idade pós-natal mediana de 4,8 meses.94 A taxa de sobrevivência desta amostra foi de 64% aos seis meses após o diagnóstico de hipertensão pulmonar. Nesta amostra, a hipertensão pulmonar severa foi um factor de risco significativo de mortalidade. É importante reconhecer que a hipertensão pulmonar pode desenvolver-se no pré-termo com displasia broncopulmonar após a alta da unidade de cuidados intensivos neonatais. As terapêuticas com NOi e sildenafil têm demonstrado benefício nestes doentes. Embora a segurança do sildenafil a longo prazo ainda não esteja estabelecida, este fármaco tem sido utilizado por períodos de um a dois anos, em casos isolados, sem efeitos adversos.95 Um caso de severa retinopatia da prematuridade foi documentado num recém-nascido prétermo tratado com sildenafil às 29 semanas de idade gestacional, levantando questões quanto à segurança deste fármaco durante o período de vulnerabilidade para desenvolvimento de retinopatia da prematuridade.96 Permanece desconhecido se o NOi ou o sildenafil melhoram a sobrevida e estimulam a angiogénese e crescimento pulmonar em crianças com displasia broncopulmonar, embora estes efeitos benéficos tenham sido documentados em modelos animais.97
Prognóstico
A taxa de sobrevivência na HPPRN é superior a 90%. No entanto, existe considerável diferença na sobrevivência e sequelas a longo prazo na dependência da causa da HPPRN.3
Vários estudos avaliaram o neurodesenvolvimento de sobreviventes de HPPRN aos 18 – 24 meses de vida.98-101 Estes estudos identificaram significativo risco para deficiência auditiva bem como outras alterações do neurodesenvolvimento, incluindo paralisia cerebral, deficiência visual, índice de desenvolvimento mental de Bayley ou índice de desenvolvimento psicomotor inferior a 70 e exame neurológico anormal.
Estes dados demonstram a necessidade de seguimento a longo prazo destes doentes.
Referências
1. Gersony WM, Duc GV, Sinclair JC. PFC syndrome (persistence of the fetal circulation). Circulation 1969; 40 (Suppl 3): 87. [ Links ]
2. Lucas VW, Ginsberg H. Persistent pulmonary hypertension of the newborn. In: Goldsmith JP, Karotkin EH, editors. Assisted Ventilation of the Neonate. 4th edition. Philadelphia: Saunders; 2003, p 398 – 403. [ Links ]
3. Konduri GG, Kim UO. Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn. Pediatr Clin North Am 2009; 56: 579 – 600. [ Links ]
4. Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, Stevenson DK, et al. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics 2000; 105: 14 – 20. [ Links ]
5. Danhaive O, Margossian R, Geva T, Kourembanas S. Pulmonary hypertension and right ventricular dysfunction in growth-restricted, extremely low birth weight neonates. J Perinatol 2005; 25: 495 – 9. [ Links ]
6. Rothstein R, Paris Y, Quizon A. Pulmonary hypertension. Pediatrics in Review 2009; 30: 39 - 46 [ Links ]
7. Proceedings of the 3rd World Symposium on Pulmonary Arterial Hypertension. Venice, Italy, June 23 – 25, 2003. J Am Coll Cardiol 2004; 43 (suppl 12): 1S – 90S. [ Links ]
8. Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, et al. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics 2004; 113: 559 – 64. [ Links ]
9. Yoder BA, Kirsch EA, Barth WH, Gordon MC. Changing obstetric practices associated with decreasing incidence of meconium aspiration syndrome. Obstet Gynecol 2002; 99: 731 – 9. [ Links ]
10. Dawes GS, Mott JC, Widdicombe JG, Wyatt DG. Changes in the lungs of the newborn lamb. J Physiol 1953; 121: 141 – 62. [ Links ]
11. Lakshminrisimha S, Steinhorn RH. Pulmonary vascular biology during neonatal transition. Clin Perinatol 1999; 26: 601 – 19. [ Links ]
12. Rasanen J, Wood DC, Debbs RH, Cohen J, Weiner S, Huhta JC. Reactivity of the human fetal pulmonary circulation to maternal oxygenation increases during the second half of pregnancy: a randomized study. Circulation1998; 97: 257 – 62. [ Links ]
13. Morin FC 3rd, Egan EA, Ferguson W, Lundgren CE. Development of pulmonary vascular response to oxygen. Am J Physiol 1988; 254: H542 – 6. [ Links ]
14. Cassin S, Dawes GS, Mott GC, Ross BB, Strang LB. The vascular resistance of the foetal and newly ventilated lung of the lamb. J Physiol 1964; 171: 61 – 79. [ Links ]
15. Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol 1990; 259: H 1921 – 7. [ Links ]
16. Tiktinsky MH, Morin FC III. Increasing oxygen tension dilates fetal pulmonary circulation via endothelium-derived relaxing factor. Am J Physiol 1993; 265 (1 Pt 2): H 376 – 80. [ Links ]
17. Shaul PW, Wells LB. Oxygen modulates nitric oxide production selectively in fetal pulmonary endothelial cells. Am J Respir Cell Mol Biol 1994; 11: 432 – 8. [ Links ]
18. Shaul PW, Campbell WB, Farrar MA, Magness RR. Oxygen modulates prostacyclin synthesis in ovine fetal pulmonary arteries by an effect on cyclooxigenase. J Clin Invest 1992; 90: 2147 – 55. [ Links ]
19. Konduri GG, Mattei J. Role of oxidative phospholylation and ATP release in birth related pulmonary vasodilation in fetal lambs. Am J Physiol Heart Circ Physiol 2002; 283: H 1600 – 8. [ Links ]
20. Konduri GG, Mital S, Gervasio CT, Rotta AT, Forman K. Purine nucleotides contribute to pulmonary vasodilation caused by birth related stimuli in the ovine fetus. (Heart Circ Physiol 41). Am J Physiol 1997; 272: H 2377 – 84. [ Links ]
21. Shaul PW, Farrar, MA, Magness RR. Pulmonary endothelial nitric oxide production is developmentally regulated in the fetus and newborn. Am J Physiol 1993; 265 Heart Circ Physiol 34): H1056 – 63. [ Links ]
22. Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH.. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signalling. Am J Respir Crit Care Med 2007; 176: 1146 – 53. [ Links ]
23. Grover TR, Parker TA, Markham NE, Abman SH. rhVEGF treatment preserves pulmonary vascular reactivity and structure in na experimental model of pulmonary hypertension in fetal sheep. Am J Physiol Lung Cell Mol Physiol 2005; 289: L 315 – 21. [ Links ]
24. Matsusshita T, Hislopp AA, Boels PJ, Deutsch J, Haworth SG. Changes in ANP responsiveness of normal and hypertensive porcine intrapulmonary arteries during maturation. Pediatr Res 1999; 46: 411 – 8. [ Links ]
25. Leffler CW, Hessler JR, Green RS. The onset of breathing at birth stimulates pulmonary ascular prostacyclin synthesis. Pediatr Res 1984; 18: 938 – 42. [ Links ]
26. Villanueva ME, Zaher FM, Svinarich DM, Konduri GG. Decreased gene expression of endothelial nitric oxide synthase in newborns with persistent pulmonary hypertension. Pediatr Res 1998;44: 338 – 43. [ Links ]
27. Pearson DL, Dawling S, Walsh W, Haines JL, Christman BW, Bazyk A, et al. Neonatal pulmonary hypertension: urea-cycle intermediates, nitric oxide production and carbarnoylphosphate syntthetase function. N Engl J Med 2001; 344: 1832 – 8. [ Links ]
28. Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC 3rd. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol 1997; 272 (Lung Cell Mol Physiol): L 1005 – 12. [ Links ]
29. Villamor E, Le Cras TD, Horan MP, Halbower AC, Tuder RM, Abman SH. Chronic intrauterine pulmonary hypertension impairs endothelial nitric oxide synthase in the ovine fetus. Am J Physiol 1997; 272 (Lung Cell Mol Physiol): L 1013 – 20. [ Links ]
30. Arrigoni FL, Vallance P, Haworth SG. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation 2003; 107: 1195 – 201. [ Links ]
31. Pullamsetti S, Kiss L, Ghofrani HA, Voswinckel R, Haredza P, Klepetko W, et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J 2005; 19: 1175 – 7. [ Links ]
32. Rosenberg AA, Kennaugh J, Koppenhaffer SL, Loomis M, Chatfield BA, Abman SH.Elevated immunoreactive endothelin-1 levels in newborn infants with persistent pulmonary hypertension. J Pediatr 1994; 124: 489 – 90. [ Links ]
33. Wedgwood S, Black SM. Endothelin-1 decreases endothelial NOS expression and activity through ETA receptor-mediated generation of hydrogen peroxide. Am J Physiol Lung Cell Mol Physiol 2005; 288: l 480 – 7. [ Links ]
34. Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, et al. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Cir Res 2003; 92: 683 – 91. [ Links ]
35. Konduri GG, Bakhutashvili I, Eis A, Pritchard K Jr. Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 2007; 292: H 1812 – 20. [ Links ]
36. Tzao C, Nickerson PA, Russell JA, Gugino SF, Steinhorn RH. Pulmonary hypertension alters soluble guanylate cyclase activity and expression in pulmonary arteries isolated from fetal lambs. Pediatr Pulmonol 2001; 31: 97 – 105. [ Links ]
37. Csaba IF, Sulyok E, Ertl T. Relationship of maternal treatment with indomethacin to persistence of fetal circulation syndrome. J Pediatr 1978; 92: 484. [ Links ]
38. Rubaltelli FF, Chiozza ML, Zanardo V, Cantarutti F. Effect on neonate of maternal treatment with indomethacin. J Pediatr 1979; 94: 161. [ Links ]
39. Alano MA, Ngougmna E, Ostrea EM, Konduri GG. Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Pediatrics 2001; 107: 519 – 23. [ Links ]
40. Abman SH, Shanley PF, Accurso F. Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest 1989; 83: 1849 – 58. [ Links ]
41. Morin FC. Ligating the dctus arteriosus before birth causes persistent pulmonary hypertension in newborn lamb. Pediatr Res 1989; 25: 245 – 50.26 [ Links ]
42. Grover TR, Parker TA, Balasubramaniam V, Markham NE, Abman SH. Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2005; 288: L 648 – 54. [ Links ]
43. Chambers CD, Hernandez-Dias S, Van Mater LJ, Werler MM, Louik C, Jones KL, et al. Selective serotonin-re-uptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 2006; 354; 579 – 87. [ Links ]
44. Andrade SE, Mc Phillips H, Loren D, Raebel MA, Lane K, Livingston J, et al. Antidepressant medication use and risk of persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf 2009; 18: 246 – 52. [ Links ]
45. Consensus statement on the management of pulmonary hypertension in clinical practice in the UK and Ireland. National Pulmonary hypertension centres of the UK and Ireland. Thorax 2008; 63:ii1 – ii41 [ Links ]
46. O`Donovan D, McMahon C, Costigan C, Oslizlok P, Duff D. Reversible pulmonary hypertension in neonatal Graves disease. Ir Med J 1997; 90: 147 – 8. [ Links ]
47. Zimmerman D. Fetal and neonatal hyperthyroidism. Thyroid 1999; 9: 727 – 33. [ Links ]
48. Oden J, Cheifetz IM. Neonatal thyrotoxicosis and persistent pulmonary hypertension necessitating extracorporeal life support. Pediatrics 2005; 115: e105 – 8. [ Links ]
49. Maquet E, Costagliola S, Parma J, Christophe-Hobertus C, Oligny L, Fournet JC, e tal. Lethal respiratory failure and mild primary hypothyroidism in a term girl with a de Novo heterozygous mutation in the TITF1/NKX2.1 gene. J Clin Endocrinol Metab 2009; 94: 197 – 203.
50. Mukerjee D, Coleiro B, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003; 62: 1088 – 93. [ Links ]
51. Morse JH, Barst RJ, Whitman HH 3rd, Fotino M, Jacobs JC. Isolated pulmonary hypertension in the grandchild of a kindred with scleroderma (systemic sclerosis): neonatal scleroderma? J Rheumatol 1989; 16: 1536 – 41. [ Links ]
52. Degano B, Sitbon O, Simonneau G. Pulmonary arterial hypertension and HIV infection. Semin Respir Crit Care Med 2009; 30: 440 – 7. [ Links ]
53. Feiterna-Sperling C, Huseman D, Timme J, Buhrer C, Obladen M. Resolution of human immunodeficiency virus type 1 infection-related severe pulmonary hypertension in a low-birth-weight infant. Pediatr Infect Dis J 2008; 27: 564 – 7. [ Links ]
54. Rich S, Dantzker, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med 1987; 107: 216 – 23. [ Links ]
55. Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-q32. Nat Genet 1997; 15: 277 80. [ Links ]
56. Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 1997; 95: 2603 – 6. [ Links ]
57. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor.II gene. AmJ Hum Genet 2000; 67: 737 – 744. [ Links ]
58. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension: The International PPH Consortium. Nat Genet 2000; 26: 81 – 4. [ Links ]
59. Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA 3rd, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 2001; 345: 319 – 24. [ Links ]
60. Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 2003; 40: 865 – 71. [ Links ]
61. Hawkins A, Tulloh R. Treatment of pediatric pulmonary hypertension. Vascular Health and Risk Management 2009; 5: 509 – 24. [ Links ]
62. Cheung PY, Tyebkhan JM, Peliowski, Ainsworth W, Robertson CM. Prolonged use of pancuronium bromide and sensorioneural hearing loss in childhood survivors of congenital diaphragmatic hernia. J Pediatr 1999; 135: 233 – 9. [ Links ]
63. Robertson C, Tyberkhan JM, Peliowski A, Etches PC, Cheung PY. Ototoxic drugs and sensorioneural hearing loss following severe neonatal respiratory failure. Acta Paediatr 2006; 95: 214 – 23. [ Links ]
64. Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology 2007; 91: 283 – 90. [ Links ]
65. Schreiber MD, Heymann MA, Soifer SJ. Increased arterial pH, not decreased PaCO2, attenuates hypoxia-induced pulmonary vasoconstriction in newborn lambs. Pediatr Res 1986; 20: 113 – 7. [ Links ]
66. Hendricks-Munoz KD, Walton JP. Hearing loss in infants with persistent fetal circulation. Pediatrics 1988; 81: 650 – 6. [ Links ]
67. Marron MJ, Crisafi MA, Driscoll JM Jr, Wung JT, Driscol YT, Fay TH et al. Hearing loss and neurodevelopmental outcome in survivors of persistent pulmonary hypertension of the newborn. Pediatrics 1992: 90: 392 – 6. [ Links ]
68. Lotze A, Mitchell BR, Bulas DI, Zola EM, Shalwitt RA, Gunkel JH. Multicenter study of surfactant (beractant) use in the treatment of term infants with severe respiratory failure. J Pediatr 1998; 132: 40 – 7. [ Links ]
69. Tourneux P, Rakza T, Bouissou A, Krim G, Storme L. Pulmonary circulatory effects of norepinhphrine in newborn infants with persistent pulmonary hypertension. J Pediatr 2008; 153: 345 – 9. [ Links ]
70. Gonçalves G, Birne A, Chaves F. Hipertensão pulmonar e terapêutica com óxido nítrico. Consensos Nacionais em Neonatologia. Secção de Neonatologia da Sociedade Portuguesa de Pediatria. Angelini Farmacêutica. Coimbra 2004: 89 - 95 [ Links ]
71. American Academy of Pediatrics Committee on Fetus and Newborn. Use of inhaled nitric oxide. Pediatrics 2000; 106: 344 – 5. [ Links ]
72. The Neonatal Inhaled Nitric Oxide Study Group. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. N Engl J Med 1997; 336: 597 – 604. [ Links ]
73. Roberts JD, Fineman JR, Morin FC 3rd, Shaul PW, Rimar S, Shreiber MD et al. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. N Engl J Med 1997; 336: 605 – 10. [ Links ]
74. Christou H, Van Marter LJ, Wessel DL, Allred EN, Kane JW, Thompson JE et al. Inhaled nitric oxide reduces the need for extracorporeal membrane oxygenation in infants with persistent pulmonary hypertension of the newborn. Crit Care Med 2000; 28: 3722 – 7. [ Links ]
75. Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA et al. Low dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Oxide Nitric Research Group. N Engl J Med 2000; 342: 469 – 74. [ Links ]
76. Wessel DL, Adatia I, Van Marter LJ, Thompson JE, Kane JW, Stark AR et al. Improved oxygenation in a randomised trial of inhaled nitric oxide for persistent pulmonary hypetension of the newborn. Pediatrics 1997; 100: E7. [ Links ]
77. Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R et al. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double masked, placebo controled, dese-response, multicenter study. Pediatrics 1998; 101: 325 – 34. [ Links ]
78. Lakshminrusimha S, Russel JA, Steinhorm RH, Swartz DD, Ryan RM, Gugino SF et al. Pulmonary hemodynamics in neonatal lambs resuscitated with 21%, 50% and 100% oxygen. Pediatr Res 2007; 62: 313 – 8. [ Links ]
79. Lakshminrusimha S, Russel JA, Wedgwood S, Gugino SF, Kazzaz JA, Davis JM et al. Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care Med 2006; 174: 1370 – 7. [ Links ]
80. Neonatal Inhaled Nitric Oxide Study Group (NINOS). Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. Pediatrics 1997; 99: 838-45. [ Links ]
81. Atz AM, Wessel DL. Sildenafil ameliorates effects of inhaled nitric oxide withdrawal. Anesthesiology 1999; 91: 307 – 10. [ Links ]
82. Baquero H, Soliz A, Neira F, Venegas ME, Sola A. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: a pilot randomized, blinded study. Pediatrics 2006; 117: 1077 – 83. [ Links ]
83. Travadi JN, Patole SK. Phosphodiesterase inhibitors for persistent pulmonary hypertension of the newborn: a review. Pediatr Pulmonol 2003; 36: 529 – 35. [ Links ]
84. Teixeira CE, Priviero FB, Webb RC. Differential effects of the phosphodiesterase type 5 inhibitors sildenafil, verdenafil, and tadalafil in rat aorta. J Pharmacol Exp Ther 2006; 316: 654 – 61. [ Links ]
85. Toque HA, Teixeira CE, Priviero FB, Morganti RP, Antunes E, De Nucci G. Verdenafil, but not sildenafil or tadalafil, has calcium-channel blocking activity in rabbit isolated pulmonary artery and human washed platelets. Br J Pharmacol 2008; 154: 787 – 96. [ Links ]
86. Takaoka S, Faul JL, Doyle R. Current therapies for pulmonary arterial hypertension. Semin Cardiothorac Vasc Anesth 2007; 11: 137 – 48. [ Links ]
87. Kelly LK, Porta NF, Goodman DM, Carroll CL, Steinhorn RH. Inhaled prostacyclin for term infants with persistent pulmonary hypertension refractory to inhaled nitric oxide. J Pediatr 2002; 141: 830 – 2. [ Links ]
88. Chang AC, Atz AM, Wernovsky G, Burke RP, Wessel DL. Milrinone: systemic and pulmonary hemodinamic effects in neonates after cardiac surgery. Crit Care Med 1995; 23: 1907 – 14. [ Links ]
89. Bassler D, Choong K, McNamara P, Kirpalani H. Neonatal persistent pulmonary hypertension treated with milrinone: four case reports. Biol Neonate 2006; 89: 1 – 5. [ Links ]
90. McNamara PJ, Laique F, Muang-In S, Whyte HE. Milrinone improves oxygenation in neonates with severe persistent pulmonary hypertension of the newborn. J Crit Care 2006; 21: 217 – 22. [ Links ]
91. Goissen C, Ghyselen L, Tourneux P, Krim G, Storme L, Bou P, et al. Persistent pulmonary hypertension of the newborn with transposition of the great arteries: successful treatment with bosentan. Eur J Pediatr 2008; 167: 437 – 40. [ Links ]
92. Nakwan N, Choksuchat D, Saksawad R, Thammachote P, Nakwan N. Successful treatment of persistent pulmonary hypertension of the newborn with bosentan. Acta Paediatr 2009; 98: 1683 – 5. [ Links ]
93. Chandrasekar I, Eis A, Konduri GG. Betamethasone attenuates oxidant stress in endothelial cells from fetal lambs with persistent pulmonary hypertension. Pediatr Res 2008; 63: 67 – 72. [ Links ]
94. Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV, Thomas KC et al. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics 2007; 120: 1260 – 9. [ Links ]
95. Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr 2009; 154: 379 – 84. [ Links ]
96. Marsh CS, Marden B, Newsom R. Severe retinopathy of prematurity (ROP) in a premature baby treated with sildenafil acetate (Viagra) for pulmonary hypertension. Br J Ophthalmol 2004; 88: 306 – 7. [ Links ]
97. Arul N, Konduri GG. Inhaled nitric oxide for preterm neonates. Clin Perinatol 2009; 36: 43 – 61. [ Links ]
98. Konduri GG, Vohr B, Robertson C. Early inhaled nitric oxide therapy for term and near-term newborn infants with hypoxic respiratory failure: neurodevelopmental follow-up. J Pediatr 2007; 150: 235 – 40. [ Links ]
99. The Neonatal Inhaled Nitric Oxide Study Group. Inhaled nitric oxide in term and near term infants: neurodevelopmental follow-up of the Neonatal Inhaled Nitric Oxide Study Group (NINOS). J Pediatr 2000; 136: 611 – 7. [ Links ]
100. Robertson CM, Tyebkhan JM, Hagler ME, Cheung PY, Peliowski A, Etches PC. Late-onset, progressive sensorineural hearing loss after severe neonatal respiratory failure. Otol Neurotol 2002; 23: 353 – 6. [ Links ]
101. Lipkin PH, Davidson D, Spivak L, Straube R, Rhines J, Chang CT. Neurodevelopmental and medical outcomes of persistent pulmonary hypertension in term newborns treated with nitric oxide. J Pediatr 2002; 140: 306 – 10. [ Links ]
Gustavo Rocha
Serviço de Neonatologia / Departamento de Pediatria, Hospital de São João – Piso 2. Al. Prof. Hernâni Monteiro, 4200 – 319 Porto. Email: gusrocha@oninet.pt

