










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.27 no.1 Porto fev. 2013
ARTIGO DE REVISÃO
Doença celíaca refratária
Refractory coeliac disease
Cunha M1, Carneiro F1, Amil J2
1Serviço de anatomia patológica, faculdade de medicina
2Serviço de pediatria, centro hospitalar S. João, Porto
RESUMO
A Doença Celíaca (DC) tem uma patogénese complexa que resulta da interação entre fatores ambientais (exposição digestiva ao glúten) e fatores genéticos e imunológicos. O tratamento com dieta sem glúten (DSG) induz melhoria clínica e histológica na maioria dos doentes. No entanto, uma proporção não responde à DSG, correspondendo ao diagnóstico de Doença Celíaca Refratária (DCR). A DCR pode ser classificada em tipo 1, caracterizada pela presença de linfócitos intraepiteliais policlonais, e com imunofenótipo normal, e tipo 2, com expansão clonal de linfócitos intraepiteliais com imunofenótipo aberrante. A DCR está associada a complicações como má nutrição, gastrite linfocítica, jejunite ulcerativa e linfoma de células T associado a enteropatia (LCTAE), mais frequentes na DCR de tipo 2. O prognóstico da DCR de tipo 2 é reservado com uma sobrevida aos 5 anos de aproximadamente 50%. A DCR pode ser considerada uma condição precursora de LCTAE. O tratamento da DCR, nomeadamente de tipo 2, continua a constituir um desafio clínico.
Palavras-chave: Doença celíaca refratária, linfoma de células T associado à enteropatia, histopatologia
ABSTRACT
Coeliac disease has a complex pathogenesis that results from the interaction between environmental (dietary exposure to gluten), genetic and immunologic factors. Gluten-free diet induces clinical and histological recovery in the majority of patients. However, a proportion of patients does not respond to a gluten-free diet and will be diagnosed as Refractory Coeliac Disease (RCD). RCD can be classified in type 1, characterized by polyclonal intraepithelial lymphocytes with a normal immune phenotype, and type 2, characterized by monoclonal intraepithelial lymphocytes with an aberrant immune phenotype. RCD is associated with a high risk of complications such as malnutrition, lymphocytic gastritis, ulcerative jejunitis and enteropathy-associated T-cell lymphoma, especially type 2. The prognosis is poor in RCD type 2, with a 5-year survival of approximately 50%. RCD can be considered a precursor of lymphoma. The treatment of RCD, mainly type 2, remains a clinical challenge.
Key-words: Refractory celiac disease, enteropathy-type t-cell lymphoma, histopathology
INTRODUÇÃO
A Doença Celíaca (DC) é a causa mais comum de atrofia da mucosa do intestino delgado proximal e está relacionada com a intolerância ao glúten em indivíduos com suscetibilidade genética. De acordo com um único artigo publicado com dados epidemiológicos portugueses, referentes a uma amostra de 536 indivíduos, a DC foi identificada em apenas 4 indivíduos (1 em cada 134), o que provavelmente traduz uma infra estimação da frequência desta doença em Portugal que de acordo com dados internacionais pode chegar a 1 em 100 indivíduos.1 A dieta sem glúten (DSG) induz melhoria clínica significativa no intervalo de dias a semanas. No entanto, na maior parte dos doentes celíacos, a recuperação morfológica da mucosa para um padrão histológico normal pode demorar meses e até anos e tende a ser mais rápida em crianças do que em adultos.2,3 Em 10% a 17% dos doentes a DSG não induz melhoria clínica ou histológica.2,4 Estes dados sugerem que a patogenia da doença é complexa e tem-se apresentado explicações para esta situação: contaminação com glúten dos alimentos (principal causa) e a Doença Celíaca Refractária (DCR), que é responsável por 7% a 18% destes casos.2,3 No entanto, para se estabelecer o diagnóstico de DCR é necessário excluir outras doenças que podem causar diarreia crónica, má absorção e/ou atrofia vilositária. Os critérios de diagnóstico de DCR consistem em: i) persistência de alterações morfológicas da mucosa intestinal (atrofia vilositária; hiperplasia das criptas e linfocitose intraepitelial) após 6-12 meses de DSG; ii) persistência de sintomas graves com necessidade de intervenção clínica, independentemente da duração da DSG.3,5,6 A DCR pode ter apresentação primária ou secundária definidas pela existência ou não de uma resposta inicial transitória à DSG.7 A DCR é comumente associada exclusivamente a adultos, mas foi recentemente publicado um caso de DCR secundária numa criança.7
MATERIAL E MÉTODOS
Pesquisa bibliográfica
A pesquisa bibliográfica foi levada a cabo recorrendo à base de dados online da pubmed/MEDLiNE, http://www.ncbi.nlm.nih.gov/pubmed, foi empregue o seguinte descritor: refractory coeliac disease. A pesquisa devolveu 315 artigos indexados, publica-dos até Dezembro de 2011.
Utilizaram-se também livros de texto e conteúdos online referenciados como artigos de revisão.
Critérios de elegibilidade para esta revisão
Para compilar o máximo de informação foram considerados todos os abstracts referentes à pesquisa mencionada e selecionados os artigos para leitura integral.
Para a seleção utilizaram-se os seguintes critérios de inclusão:(1)artigos publicados em língua inglesa ou portuguesa; (2) todos os estudos referentes a Doença CelíacaRefratária; (3) todos os artigos de autores/ grupos de investigação com várias publicações neste domínio foram incluídos. Foram excluídos todos os seguintes: (1) sem acesso a texto integral; (2) dados clínico-patológicos omissos ou inapropriados; (3) metanálises ou revisões sistemáticas sem dados originais; (4) tópicos fora do âmbito temático desta revisão.
PATOGÉNESE
A Doença Celíaca (DC) tem patogenia complexa que resulta da interação entre fatores ambientais (nomeadamente o glúten), fatores genéticos e fatores imunológicos. A DC é induzida pela ingestão de glúten que existe no trigo, na cevada e no centeio.8 Recentemente foi identificada a intolerância à aveia em alguns indivíduos com DC.9
O papel do glúten
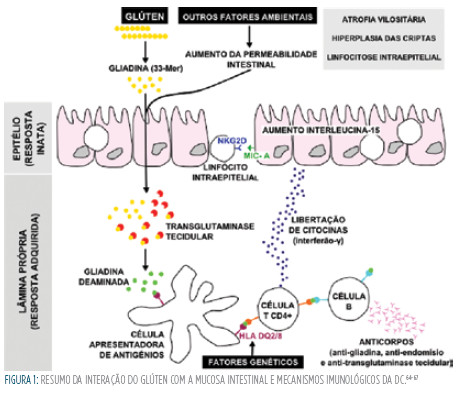
A proteína do glúten tem elevado teor de glutamina e prolina e é pouco digerida no trato gastrointestinal superior. A gliadina constitui a fração do glúten, que é solúvel em álcool e que contém a maior parte dos componentes tóxicos para a mucosa.8 As moléculas de gliadina não digeridas (nomeadamente um péptido da fração a-gliadina composto por 33 aminoácidos, 33-mer) são resistentes à degradação pelos sucos gástrico, pancreático e intestinal.8 Assim, estas moléculas de gliadina permanecem no lúmen intestinal após a degradação do glúten.8 A abundância e a localização dos resíduos de prolina são apontadas como uma das razões para esta resistência.8 A prolina protege os péptidos da proteólise, determina a especificidade da transglutaminase tecidular e é crucial para a ligação às moléculas HLA-DQ2.9 é por isso previsível que a utilização de uma endopeptidase de prolina possa diminuir a toxicidade do glúten nestes doentes.8 A glutamina e a prolina atravessam a barreira do epitélio intestinal, provavelmente no decurso de infeções ou quando há aumento da permeabilidade por outras causas. No córion da mucosa, estas moléculas interatuam com as células apresentadoras de antigénios.8
As aveninas (presentes na aveia) provocam intolerância em alguns doentes com DC.9 Apesar de terem cerca de metade dos resíduos de prolina presentes no trigo, cevada e centeio, os epítopos de avenina localizam-se em regiões análogas às da gliadina e têm elevado teor de prolina e glutamina.9
Resposta imune da mucosa
Nos doentes celíacos a resposta imune às frações de gliadina promove uma reação inflamatória, no intestino proximal, caracterizada por infiltração do epitélio e lâmina própria por células inflamatórias crónicas e atrofia vilositária. Esta resposta é mediada pelo sistema imune inato e adquirido.10 A resposta adquirida ocorre primariamente na lâmina própria após a entrada dos péptidos de gliadina para este espaço por aumento da permeabilidade da mucosa. A resposta inata ocorre predominantemente no epitélio intestinal.
A resposta do sistema imune adquirido é mediada pelas células T CD4+ reativas à gliadina presentes na lâmina própria que reconhecem os seus péptidos.10 As células T ligam-se às moléculas HLA da classe ii, DQ2 ou DQ8, nas células apresentadoras de antigénios.10 Consequentemente, as células T produzem citocinas pró-inflamatórias, particularmente o interferão-?.10,11 Esta citocina encontra-se particularmente aumentada na DC não tratada e na DCR.12
A transglutaminase tecidular é uma enzima que desamina os peptídeos de gliadina, aumentando, assim, a sua imunogenicidade.10 A cascata de reações inflamatórias condiciona a libertação de metaloproteinases (mmp-1, mmp-3, mmp-9 e inibidor tecidular das metapoproteinases 1 – Timp-1) e de outros mediadores que lesam a mucosa intestinal induzindo a hiperplasia das criptas, a atrofia vilositária e ativação das células Bque produzem anticorpos.13 As metaloproteinases estão mais aumentadas em biopsias com lesões mais graves sugerindo um papel destas enzimas na constituição das lesões.13
A resposta do sistema imune inato é caracterizada por um aumento do sistema da interleucina 15 nos enterócitos, que aumenta os linfócitos intraepiteliais que expressam o recetor NK-G2D, um marcador de células Natural Killer.14 Estas células ativadas tornam-se citotóxicas e destroem os enterócitos que expressam a cadeia A do complexo major de histocompatibilidade da classe i (MIC-A), um antigénio da superfície das células epiteliais que é induzido pelainterleucina15 em situações de stress como nas infeções (Figura 1).15,16 Na DCR há uma expressão aumentada e incontrolada da interleucina 15, perpetuando a ativação dos linfócitos intraepiteliais resultando na proliferação clonal dos linfócitos T e emissão de sinais anti-apoptóticos.14,17,18 No entanto, o número de células Natural Killer na DCR é inferior ao encontrado na DC com DSG.19 No linfoma de células T associado a enteropatia (LCTAE) os níveis destas células são também muito baixos e comparáveis aos da DCR.19 Com base nesta evidência é provável que bloqueadores desta interleucina ou fármacos que promovam o aumento de células Natural Killer possam vir a revelar-se com potencial terapêutico nestes doentes. A utilização de modelos animais (primatas, por exemplo) constitui uma possibilidade para a investigação dos mecanismos fisiopatológicos desta doença e para a identificação de futuras armas terapêuticas.20
Fatores genéticos
A existência destes fatores é comprovada pela prevalência aumentada (13%) em familiares de primeiro grau.21,22 A DC desenvolve-se quase exclusivamente em indivíduos portadores dos alelos que codificam as proteínas HLA-DQ2 ou HLA-DQ8.23 No entanto, estes alelos estão também presentes em indivíduos que não apresentam DC. Assim, a sua presença é necessária mas não suficiente para o desenvolvimento de DC.22 Vários genes não relacionados com o HLA foram identificados mas a sua influência na DC não foi comprovada. 10 Constitui um exemplo a molécula Cytotoxic T-Lymphocyte Antigen 4 (CTLA4).10
DIAGNÓSTICO E CLASSIFICAÇÃO
A Doença Celíaca Refratária (DCR) pode ser definida segundo os seguintes critérios: (1) recorrência ou persistência dos sintomas (diarreia, perda de peso involuntária e/ou dor abdominal) e lesão intestinal (pelo menos atrofia vilositária parcial) após DSG durante pelo menos 6-12 meses6; (2) exclusão de outras causas de DC sem resposta à DSG, tais como contaminação intencional ou inadvertida com glúten6; (3) necessidade de terapia alternativa por perda de resposta à dieta sem glúten (DSG)6; (4) ausência de linfoma intestinal ou sistémico6; (5) diagnóstico prévio comprovado por biópsia com história de resposta clínica à DSG, testes serológicos positivos, presença de HLA-DQ2 ou HLA-DQ8 e história familiar de Doença Celíaca (DC); (6) subtipos de DCR identificados pela ausência (DCR de tipo 1) ou presença (DCR de tipo 2) de um fenótipo aberrante (monoclonal) de linfócitos intraepiteliais.6 A presença de todos os critérios permite diagnosticar com certeza como DCR.6
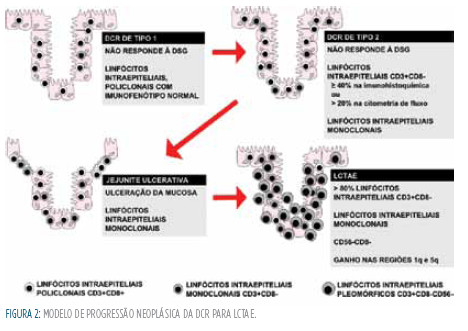
ADCRpodeserclassificadaemdoistipos:tipo1,em que os linfócitos intraepiteliais são policlonais e têm um fenótipo normal; e tipo 2, com expansão clonal de uma população de linfócitos intraepiteliais com imunofenótipo aberrante (=40% CD3+CD8-detetados por imunocitoquímica ou > 20% por citometria de fluxo,e antigénios do recetor de células T).24,25
Quando o paciente se apresenta com diarreia crónica (definida como =3 dejeções por dia durante pelo menos 6 meses) refratária à DSG devem excluir-se outras causas.4 Estas dividem-se em quatro categorias:alteraçãodafunção gastrointestinalsem alterações histopatológicas (Síndrome do intestino irritável ou disfunção do esfíncter anal); má absorção de carbo-hidratos causada por ingestão aumentada de frutoseouintolerânciaàlactose;máabsorçãodenutrientes causada por insuficiência do pâncreas exócrino; má absorção de fluidos e eletrólitos causada por colite microscópica.4
A endoscopia com biópsia duodenal permite a confirmação do diagnóstico de DC.26 marsh27 propôs uma classificação histológica em 1992 que foi modificada por Oberhuber28 e simplificada recentemente por Corazza29. A escolha da classificação depende do patologista e do clínico.26 Neste trabalho apresenta-se a classificação de Oberhuber ou marsh modificada por ser a que mais vezes é referenciada: tipo 0 – mucosa normal; tipo 1 – aumento dos linfócitos intraepiteliais, mucosa de arquitetura normal e altura normal das criptas; tipo 2 – vilosidades de arquitetura normal, aumento dos linfócitos intraepiteliais e criptas hiperplásicas; tipo 3 – tipo destrutivo com vários graus de atrofia vilositária, em todos os subtipos há aumento da altura das criptas e presença de células inflamatórias; subtipo 3a – atrofia vilositária parcial, vilosidades alargadas e encurtadas com um ratio vilosidade/ cripta de 1:1; subtipo 3b – atrofia vilositária subtotal, vilosidades atróficas mas ainda separadas e identificáveis; subtipo 3c – atrofia vilositária total, vilosidades rudimentares ou ausentes, mucosa semelhante a cólon; tipo 4 – lesão atrófica e hipoplásica, mucosa plana com criptas de altura normal, sem inflamação significativa e com contagem de linfócitos intraepiteliais normal.26,28
Foi proposto recentemente um sistema de classificação baseado em cinco fatores de prognóstico: albumina =3,2g/dL, hemoglobina =11g/dL, idade =65 anos, presença de linfócitos intraepiteliais com fenótipo anormal e lesão histológica (3c de marsh modificado).6 De acordo com estes fatores, foram definidas três categorias: i (nenhum ou 1 fator de risco) – sobrevida aos 5 anos: 96%; ii (2 ou 3) – sobrevida aos 5 anos: 71%; iii (=4) – sobrevida aos 5 anos: 19%.6 Este sistema foi utilizado num único estudo e necessita de validação. Esta classificação terá interesse para direcionar a terapêutica mediante a gravidade do prognóstico.
EXAMES COMPLEMENTARES DE DIAGNÓSTICO
Laboratoriais
Os testes serológicos utilizados para diagnóstico da DC baseiam-se na deteção de anticorpos da classe das igA como anti-gliadina, anti-endomísio e antitransglutaminase, cuja positividade confirma o diagnóstico. Uma minoria dos doentes com DCR (19%) apresenta serologia positiva persistente.6 Não há uma explicação segura para esta situação, mas foram apresentadas as seguintes interpretações: i) aumento de expressão de transglutaminase tecidular como resposta a um estado inflamatório intenso; ii) cinética anormal dos auto-anticorpos usualmente detetados na DC; iii) contaminação da dieta por baixas doses de glúten, de origem não identificada.6 Num estudo recente, foram detetados níveis elevados de anticorpos anti-calreticulina no soro de 12/13 doentes (92%) com DCR.30 De acordo com os resultados deste estudo a utilização de níveis séricos de anticorpos anti-calreticulina tem maior acuidade diagnóstica do que a utilização de anticorpos anti-gliadina, anti-endomísio e anti-transglutaminase tecidular.30
Técnicas de imagem
A enteroscopia de propulsão com biópsia jejunal foi apontada por Cellier et al como tendo valor diagnóstico para a identificação jejunite ulcerativa em doentes com DCR.31 A enteroscopia com duplo balão pode detetar ou excluir a presença de complicações de DCR como a jejunite ulcerativa e linfoma de células T associado à enteropatia (LCTAE), pois estas lesões são mais frequentes no jejuno e íleo e esta técnica permite uma observação das áreas mais distais do intestino delgado.32 Esta técnica deve ser reservada para doentes com DCR ou história de LCTAE.32
A deteção de LCTAE por técnicas de imagem (como a tomografia axial computorizada -TAC) em doentes com DCR é muitas vezes limitada por as alterações imagiológicas se poderem restringir à mucosa do intestino delgado apesar do linfoma se encontrarem estadiomaisavançado.33 A TAC pode ser uma técnica útil para o diagnóstico diferencial entre DC, LCTAE e DCR.34 Nas situações de DCR tipo 2 e LCTAE as alterações identificadas por TAC (espessamento da parede, linfadenopatia, invaginação, redução da vascularização mesentérica e baço de menores dimensões) são mais acentuadas do que na DC e DCR tipo 1.34 A tomografia com emissão depositrões (pET) usando fluor odeoxiglucose (18ffDG) revelou-se mais sensível do que a TAC para o diagnóstico de LCTAE em indivíduos com DCR.33
Avaliação do fenótipo dos linfócitos intraepiteliais
Uma razão CD8/CD3 diminuída parece ser um marcador específico de DCR, pois no epitélio, em indivíduos normais ou com DC não complicada, o CD8 é co-expresso na quase totalidade da população de linfócitos intraepiteliais.35,36 Todavia, podem existir CD3+ e CD8+, imunofenótipos normais, em indivíduos que têm DCR.37 Um imunofenótipo aberrante dos linfócitos intraepiteliais pode ser demonstrado pela razão CD8/CD3 determinada por imunocitoquímica e a monoclonalidade dos rearranjos do gene do recetor de células T pode ser determinada por polymerase Chain Reaction (pCR).24 Contudo, a presença de imunofenótipos aberrantes e a monoclonalidade do gene do recetor de células T dos linfócitos intraepitelias não são específicas da DCR, pois também são identificados na DC, embora transitoriamente, geralmente de-vido à má adesão à DSG e só muito raramente as duas alterações estão presentes em simultâneo.24 Na DCR, os imunofenótipos aberrantes e a monoclonalidade são quase sempre persistentes e existem concomitantemente.24 A presença de imunofenótipos aberrantes (=80% da população de linfócitos intraepiteliais de tipo CD3e+CD8-por imunoci-toquímica) e de monoclonalidade nas biópsias de doentes com DCR associa-se a risco aumentado de desenvolvimento de LCTAE.24 O aparecimento de alterações nos linfócitos intraepiteliais é um processo progressivo e cumulativo e uma grande proporção dos casos não apresenta estas alterações na altura do diagnóstico de DCR.24 Nos doentes com DC o aparecimento destas características nos linfócitos intraepiteliais, após confirmada a correta adesão a uma DSG, pode levantar a hipótese de DCR ou LCTAE.24 Deste modo, seria importante uma monitorização do imunofenótipo e da clonalidade dos linfócitos intraepiteliais nos doentes com DC e DCR.
Na DC os linfócitos intraepiteliais com recetores para células T γδ estão aumentados.38 um trabalho descreve, por contraste, diminuição destes linfócitos em doentes com DCR.38 Assim, a avaliação quantitativa dos linfócitos células T γδ pode constituir método de diagnóstico dos tipos de DCR, um marcador da progressão da doença e um meio para avaliação da eficácia da estratégia terapêutica na DCR tipo 2.38
COMPLICAÇÕES E PROGNÓSTICO
A Doença Celíaca Refratária (DCR) de tipo 2 está associada a pior prognóstico com ocorrência de LCTAE, má nutrição severa, frequência elevada de casos de gastrite linfocítica e jejunite ulcerativa.35,39 Todavia, embora raramente, alguns doentes classificados como DCR de tipo 1 também podem desenvolver estas complicações, nomeadamente o LCTAE que, num estudo39, foi diagnosticado em 2 de 14 doentes com DCR tipo 1.35,39 O maior número de casos com jejunite ulcerativa em doentes com DCR de tipo 2 pode explicar a maior perda proteica e subsequente má nutrição comparativamente com os de tipo 1.39 A taxa de sobrevida aos cinco anos dos doentes com DCR de tipo 2 (44%-58%) é inferior à da DCR de tipo 1 (90%-96%).39-41 Embora a principal causa de mortalidade seja a progressão para LCTAE, associada a má nutrição, as complicações trombóticas (agravadas pela inflamação e pela hipoalbuminemia) também contribuem para a elevada mortalidade nos indivíduos com DCR de tipo 2.39 A associação entre a exposição crónica ao glúten ao longo da vida e o desenvolvimento de LCTAE pode ser comprovada pela elevada frequência de resistência primária à DSG (48% na DCR de tipo2) e o diagnóstico de DCR tardio(em média em indivíduos com mais de 40 anos).39 Apesar das diferenças metodológicas e diagnósticas, estudos recentes apontam para uma maior prevalência de DCR de tipo 2 na Europa (28-75%) do que nos Estados unidos da América (14,7%).39-42 uma das possíveis razões apresentadas para este facto é a dieta com elevado teor em glúten.43
O linfoma T do intestino delgado pode ser classificado em dois subtipos: o tipo 1 está associado à DC (LCTAE) e caracteriza-se por uma população de linfócitos com variabilidade morfológica (pleomórfica, anaplástica ou imunoblástica), com negatividade para os recetores CD8 e CD56 e ganho preferente das regiões 1q e 5q, sem envolver o oncogene MYC.44 O tipo 2 ocorre esporadicamente, sem fatores de risco da DC e é constituído células neoplásicas monomórficas de pequenas dimensões, com positividade para os recetores CD8 e CD56, ganho do locus do oncogene MYC e, raramente, das regiões 1q e 5q.44,45
A proliferação clonal dos linfócitos intraepiteliais representa uma manifestação precoce de LCTAE, tal como verificado no estudo de biopsias duodenais e jejunais de indivíduos com DCR e jejunite ulcerativa, em que se identificaram linfócitos intraepiteliais monoclonais, sugerindo que estas alterações podemserconsideradasprecursorasdeLCTAE.45,46 foram encontrados linfócitos T aberrantes na camada subepitelial da mucosa intestinal, na lâmina própria, no cólon, no estômago e em localização extra-gastrointestinal como a pele, sangue, medula óssea, fígado, pulmão, gânglios linfáticos mesentéricos e seio maxilar em indivíduos com o diagnóstico de DCR de tipo 2.39,47,48 Estas observações levaram à hipótese da DCR poder ser uma doença sistémica que pode acompanhar-se de desenvolvimento de LCTAE em localização extra-intestinal.47 (Figura 2)
TRATAMENTO
O tratamento de eleição da DC é a dieta sem glúten (DSG). Este é seguro e eficaz na maior parte dos doentes, embora requeira considerável motivação dos doentes por modificar alguns hábitos alimentares tradicionais. Contudo, há doentes em que a DSG não acarreta melhorias consideráveis no quadro clínico. Por estas razões têm sido pesquisadas alternativas que complementem ou possam, no futuro, substituir a evicção de glúten. Estas têm-se focalizado em três áreas: a diminuição da exposição ao glúten, modificação da permeabilidade intestinal e modulação do sistema imune.
A utilização de corticóides (prednisolona) traduz melhorias clínicas na maioria dos doentes com qualquer tipo de DCR.6,39 No entanto, apenas 30%-40% apresentam melhoria histológica e o tratamento com corticóides sistémicos a longo prazo tem efeitos secundários.39 Um estudo com uma amostragem de apenas seis doentes com DCR demonstrou melhoria nos quatro com DCR de tipo 1 com a utilização de budesonide.49 Os dois doentes com DCR de tipo 2 não apresentaram melhoria da situação clínica.49 Noutro estudo, a terapia com budesonide em monoterapia ou em combinação com corticóides orais e/ou azatioprina permitiu obter resposta clínica em 76% dos casos (resposta completa em 55%), incluindo DCR de tipo 1 e 2.50 Num outro estudo, oito de dez doentes com DCR de tipo 1 apresentaram resposta histológica com o uso de azatioprina associada a prednisolona (para indução) e em quatro houve normalização completa da mucosa.51 Na DCR de tipo 2 esta terapêutica não permitiu obter resposta clínica ou histológica.51 Com o uso de mesalazina em dez doentes com DCR de tipo 1 em monoterapia ou em combinação com budesonide obteve-se resposta completa em 50% dos casos e 10% tiveram resposta parcial.52 Num outro estudo, a interleucina 10 não foi eficaz, na dose utilizada, num grupo de dez doentes e causou efeitos secundários, levando ao abandono da terapêutica em dois deles.53 A ciclosporina permitiu obter melhoria histológica em oito de treze indivíduos (61%) com DCR e normalização da mucosa em cinco (38%).54 A cladribina permitiu obter melhoria clínica (81%), histológica (47%) e imunológica (41%) em indivíduos com DCR de tipo 2.55 Em dois casos clínicos foi utilizado infliximab (anticorpo monoclonal anti-TNfa) como indutor da remissão com melhoria clínica dos doentes mas foi necessário fazer terapêutica de manutenção com prednisolona e/ou azatioprina.56,57 Há dois relatos de DCR com elevada percentagem de linfócitos intraepiteliais anormais em que o tratamento com alemtuzumab (anticorpo monoclonal anti-CD52) em politerapia (pred-nisona e/ou cladribina) que produziu resultados contraditórios.58,59 Num dos casos, verificou-se recuperação completa da morfologia da mucosa duodenal e redução da expressão inicialmente aumentada de CD52 nos linfócitos.58 No outro estudo, ocorreu melhoria clínica, mas a atrofia vilositária persistiu e aumentou a percentagem de linfócitos intraepiteliais anormais que continuavam a expressar CD52,59
A DCR de tipo 2 não responde completamente às terapias disponíveis e tem um risco aumentado de desenvolvimento de LCTAE. A quimioterapia em alta dose (fludarabina e melfalan) seguida de transplante de células estaminais autólogas (hematopoiéticas) produziu melhoria histológica e clínica, com diminuição da percentagem de linfócitos intraepiteliais anormais, em doentes com DCR tipo 2.60 Este estudo utilizou uma amostra pequena, com follow-up curto mas este tratamento pode revelar-se como valiosa alternativa para estes doentes.
A utilização de anticorpos que bloqueiam a interleucina 15 reduziu a lesão intestinal em ratos com expressão aumentada desta citocina nos enterócitos.61 foram testados em modelos animais derivados do dihidroisoxazole como inibidores seletivos irreversíveis da transglutaminase humana tipo 2, com resultados favoráveis.62 A ALV003 é uma pro-tease que se demonstrou ativa na degradação do glúten no estômago.63
CONCLUSÕES
A maioria dos doentes com DC responde favoravelmente à DSG. No entanto, um grupo de doentes não tem melhoria clínica e/ou histológica com persistência da sintomatologia e das alterações na mucosa intestinal após 6-12 meses de DSG, conduzindo ao diagnóstico de DCR. O diagnóstico definitivo é confirmado pelo cumprimento de critérios clínicos, analíticos e citológicos.
A imunofenotipagem permite a identificação dos sub-tipos de DCR e do risco de evolução para LCTAE (=80% da população de linfócitos intraepiteliais com fenótipo CD3+CD8-e monoclonalidade nas biopsias jejunais). A DCR, principalmente a de tipo 2, está associada a complicações como má nutrição, gastrite linfocítica, jejunite ulcerativa e LCTAE. Assim, a caracterização da DCR como de tipo 2 é importante pelo prognóstico mais sombrio (sobrevida aos 5 anos de aproximadamente 50%) e necessidade de terapêutica adequada. A via de linfomatogénese do LCTAE inicia-se na DC, evolui para DCR de tipo 1, de tipo 2 e subsequentemente jejunite ulcerativa. A DCR pode ter expressão sistémica, acompanhando-se de desenvolvimento de LCTAE em localização extra-intestinal.
A terapêutica da DCR, especialmente de tipo 2, continua a constituir um grande desafio clínico. A utilização de corticoides é eficaz na DCR de tipo 1,nomeadamente para a indução da remissão. Revelou-se também como promissora a utilização de mesalazina budesonide, cladribina e ciclosporina em casos particulares. A utilização de terapias biológicas com fármacos monoclonais pode representar um novo caminho no tratamento da DCR, em especial nas formas de maior gravidade,carecendo de validação clínica.
AGRADECIMENTO
Ao Dr. pedro Couto pela ajuda na elaboração das figuras.
REFERÊNCIAS
1. Antunes H, Abreu I, Nogueiras A, et al. [first determination of the prevalence of celiac disease in a portuguese population]. Acta med port 2006;19:115-20. [ Links ]
2. Wahab PJ, Meijer JW, Mulder CJ. Histologic follow-up of people with celiac disease on a gluten-free diet: slow and incomplete recovery. Am J Clin Pathol 2002;118:459-63. [ Links ]
3. Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: results of a systematic approach. Am J Gastroenterol 2002;97:2016-21. [ Links ]
4. Fine KD, Meyer RL, Lee EL. The prevalence and causes of chronic diarrhea in patients with celiac sprue treated with a gluten-free diet. Gastroenterology 1997;112:1830-8. [ Links ]
5. Daum S, Cellier C, Mulder CJ. Refractory coeliac disease. Best Pract Res Clin Gastroenterol 2005;19:413-24. [ Links ]
6. Rubio-Tapia A, Kelly DG, Lahr BD, Dogan A, Wu TT, Murray JA. Clinical staging and survival in refractory celiac disease: a single center experience. Gastroenterology 2009;136:99-107; quiz 352-3. [ Links ]
7. Mubarak A, Oudshoorn JH, Kneepkens CM, et al. A child with refractory coeliac disease. J Pediatr Gastroenterol Nutr 2011;53:216-8. [ Links ]
8. Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science 2002;297:2275-9. [ Links ]
9. Arentz-hansen h, fleckenstein B, molberg O, et al. The molecular basis for oat intolerance in patients with celiac disease. pLoS med 2004;1:e1. [ Links ]
10. Sollid Lm. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev immunol 2002;2:647-55. [ Links ]
11.Nilsen EM, Jahnsen FL,Lundin KE, et al. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology 1998;115:551-63. [ Links ]
12. Olaussen RW, Johansen FE, Lundin KE, Jahnsen J, Brandtzaeg P, Farstadi N. Interferon-gamma-secreting T cells localize to the epithelium in celiac disease. Scand J Immunol 2002;56:652-64. [ Links ]
13. Mohamed Bm, Feighery C, Kelly J, et al. Increased protein expression of matrix metalloproteinases -1, -3, and -9 and Timp-1 in patients with gluten-sensitive enteropathy. Dig Dis Sci 2006;51:1862-8. [ Links ]
14. Mention JJ, Ben Ahmed M, Begue B, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology 2003;125:730-45. [ Links ]
15. Meresse B, Chen Z, Ciszewski C, et al. Coordinated induction by iL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004;21:357-66. [ Links ]
16. Hue S, Mention JJ, Monteiro RC, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004;21:367-77. [ Links ]
17.Malamut G, Elmachhour R, Montcuquet N, et al. IL-15triggers an antiapoptotic pathway in human intraepitheliallymphocytes that is a potential new target in celiac disease-associated inflam¬mation and lymphomagenesis. J Clin Invest 2010;120:2131-43. [ Links ]
18. Di Sabatino A, Ciccocioppo R, Cupelli F, et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 2006;55:469-77. [ Links ]
19. Bernardo D, van hoogstraten im, Verbeek Wh, et al. Decreased circulating iNKT cell numbers in refractory coeliac disease. Clin immunol 2008;126:172-9. [ Links ]
20. Bethune MT, Borda JT, Ribka E, et al. A non-human primate model for gluten sensitivity. PLOS One 2008;3:e1614. [ Links ]
21. Bevan S, Popat S, Braegger CP, et al. Contribution of the MHC region to the familial risk of coeliac disease. J med Genet 1999;36:687-90. [ Links ]
22. Greco L, Romino R, Coto I, et al. The first large population based twin study of coeliac disease. Gut 2002;50:624-8. [ Links ]
23. Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 2005;3:843-51. [ Links ]
24. Liu H, Brais R, Lavergne-Slove A, et al. Continual monitoring of intraepithelial lymphocyte immunophenotype and clonality is more important than snapshot analysis in the surveillance of refractory coeliac disease. Gut 2010;59:452-60. [ Links ]
25.Verbeek WH, Goerresm S,Von Blomberg BM, et al. Flow cytometric determination of aberrant intra-epithelial lymphocytes predicts T-cell lymphoma development more accurately than T-cell clonality analysis in Refractory Celiac Disease. Clin Immunol 2008;126:48-56. [ Links ]
26. Walker MM, Murray JA. An update in the diagnosis of coeliac disease. Histopathology 2010. [ Links ]
27. Marshm N. Gluten, Major histo compatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity(celiacsprue).Gastroenterology 1992;102:330-54. [ Links ]
28. Oberhuber G, Granditsch G, Vogelsang h. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol hepatol 1999;11:1185-94. [ Links ]
29. Corazza GR, Villanacci V. Coeliac disease. J Clin pathol 2005;58:573-4. [ Links ]
30. Sanchez D, Palova-Jelinkova L, Felsberg J, et al. Anti-calreticulin immunoglobulin A (igA) antibodies in refractory coeliac disease. Clin Exp immunol 2008;153:351-9. [ Links ]
31. Cellier C, Cuillerier E, Patey-mariaud de Serre N, et al. Push enteroscopy in celiac sprue and refractory sprue. Gastrointest Endosc 1999;50:613-7. [ Links ]
32. Hadithi M, Al-Toma A, Oudejans J, Van Bodegraven AA, Mulder CJ, Jacobs M. The value of double-balloon enteroscopy in patients with refractory celiac disease. Am J Gastroenterol 2007;102:987-96. [ Links ]
33. Hadithi M, Mallant M, Oudejans J, Van Waesberghe JH, Mulder CJ, Comans EF. 18F-FDG PET versus CT for the detection of enteropathy-associated T-cell lymphoma in refractory celiac disease. J Nucl med 2006;47:1622-7. [ Links ]
34.Mallant M, Hadithi, Al-Toma AB, et al. Abdominal computedt omography in refractory coeliac disease and enteropathy associated T-celllymphoma. World J Gastroenterol 2007;13:1696-700. [ Links ]
35. OShea U, Abuzakouk M, Omorain C, et al. Investigation of molecular markers in the diagnosis of refractory coeliac disease in a large patient cohort. J Clin pathol 2008;61:1200-2. [ Links ]
36. Patey-Mariaud De Serre N, Cellier C, Jabri B, et al. Distinction between coeliac disease and refractory sprue: a simple immunohistochemical method. Histopathology 2000;37:70-7. [ Links ]
37. De Mascarel A, Belleannee G, Stanislas S, et al. Mucosal intraepithelial T-lymphocytes in refractory celiac disease: a neoplastic population with a variable CD8 phenotype. Am J Surg Pathol 2008;32:744-51. [ Links ]
38. Verbeek WH, Von Blomberg BM, Scholten PE, Kuik DJ, Mulder CJ, Schreurs MW. The presence of small intestinal intraepithelial gamma/delta T-lymphocytes is inversely correlated with lymphoma development in refractory celiac disease. Am J Gastroenterol 2008;103:3152-8. [ Links ]
39. Malamut G, Afchain P, Verkarre V, et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology 2009;136:81-90. [ Links ]
40. Daum S, Ipczynski R, Schumann M, Wahnschaffe U, Zeitz M, Ullrich R. High rates of complications and substantial mortality in both types of refractory sprue. Eur J Gastroenterol hepatol 2009;21:66-70. [ Links ]
41.Al-Toma A, Verbeek WH, Hadithi M, Von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy- associated T-celllymphoma: retrospective evaluation of single-centre experience. Gut 2007;56:1373-8. [ Links ]
42. Roshan B, Leffler DA, Jamma S, et al. The incidence and clinical spectrum of refractory celiac disease in a north american referral center. Am J Gastroenterol 2011;106:923-8. [ Links ]
43. Malamut G, Cellier C. Is refractory celiac disease more severe in old Europe? Am J Gastroenterol 2011;106:929-32. [ Links ]
44.Deleeuw RJ, Zettl A, Klinker E, et al. Whole-genomeanalysis and HL Agenotyping of enteropathy-type T-cell lymphoma reveals 2 distinct lymphoma subtypes. Gastroenterology 2007;132:1902-11. [ Links ]
45. Daum S, Weiss D, Hummel M, et al. Frequency of clonal intraepithelial T lymphocyte proliferations in enteropathy-type intestinal T cell lymphoma, coeliac disease, and refractory sprue. Gut 2001;49:804-12. [ Links ]
46. Bagdi E, Diss TC, Munson P, Isaacson PG. Mucosal intra-epithelial lymphocytes in enteropathy-associated T-cell lymphoma, ulcerative jejunitis, and refractory celiac disease constitute a neoplastic population. Blood 1999;94:260-4. [ Links ]
47.Verbeek WH, Von Blomberg BM, Coupe VM, Daum S, Mulder CJ, Schreursm W. Aberrant T-lymphocytes in refractory coeliac disease are not strictly confinedto a small intestinal intraepithe¬lial localization. Cytometry B Clin Cytom 2009;76:367-74. [ Links ]
48. Verkarre V, Asnafi V, Lecomte T, et al. Refractory coeliac sprue is a diffuse gastrointestinal disease. Gut 2003;52:205-11. [ Links ]
49. Daum S, Ipczynski R, Heine B, Schulzke JD, Zeitz M, Ullrich R. Therapy with budesonide in patients with refractory sprue. Digestion 2006;73:60-8. [ Links ]
50. Brar P, Lee S, Lewis S, Egbuna I, Bhagat G, Green PH. Budesonide in the treatment of refractory celiac disease. Am J Gastroenterol 2007;102:2265-9. [ Links ]
51. Goerres MS, Meijer JW, Wahab PJ, et al. Azathioprine and prednisone combination therapy in refractory coeliac disease. Aliment Pharmacol Ther 2003;18:487-94. [ Links ]
52. Jamma S, Leffler DA, Dennis m, et al. Small intestinal release mesalamine for the treatment of refractory celiac disease type i. J Clin Gastroenterol 2011;45:30-3. [ Links ]
53. mulder CJ, Wahab pJ, meijer JW, metselaar E. A pilot study of recombinant human interleukin-10 in adults with refractory coeliac disease. Eur J Gastroenterol hepatol 2001;13:1183-8. [ Links ]
54. Wahab pJ, Crusius JB, meijer JW, uil JJ, mulder CJ. Cyclosporin in the treatment of adults with refractory coeliac disease--an open pilot study. Aliment pharmacol Ther 2000;14:767-74. [ Links ]
55. Tack GJ, Verbeek WH, Al-Toma A, et al. Evaluation of Cladribine treatment in refractory celiac disease type ii. World J Gastroenterol 2011;17:506-13. [ Links ]
56. Turner SM, Moorghen M, Probert CS. Refractory coeliac disease: remission with infliximab and immunomodulators. Eur J Gastroenterol hepatol 2005;17:667-9. [ Links ]
57. Costantino G, Della Torre A, Lo Presti MA, Caruso R, Mazzon E, Fries W. Treatment of life-threatening type I refractory coeliac disease with long-term infliximab. Dig Liver Dis 2008;40:74-7. [ Links ]
58.Vivas S, Ruiz Demorales JM, Ramos F, Suarez-Vilela D. A lemtuzumab for refractory celiac disease in a patient at risk for enteropathy-associated T-cell lymphoma. N Engl J med 2006;354:2514-5. [ Links ]
59. Verbeek WH, Mulder CJ, Zweegman S. Alemtuzumab for refractory celiac disease. N Engl J med 2006;355:1396-7; author reply 7. [ Links ]
60. Al-Toma A, Visser OJ, Van Roessel HM, et al. Autologous hematopoietic stem cell transplantation in refractory celiac disease with aberrant T cells. Blood 2007;109:2243-9. [ Links ]
61. Yokoyama S, Watanabe N, Sato N, et al. Antibody-mediated blockade of iL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc Natl Acad Sci USA 2009;106:15849-54. [ Links ]
62. Choi K, Siegel M, Piper JL, et al. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol 2005;12:469-75. [ Links ]
63. Siegel M, Garber ME, Spencer AG, et al. Safety, Tolerability, and Activity of ALV003: Results from Two phase 1 Single, Escalating-Dose Clinical Trials. Dig Dis Sci 2011. [ Links ]
64. Green PH, Cellier C. Celiac disease. N Engl J Med 2007;357:1731-43. [ Links ]
65. Tjon JM, Van Bergen J, Koning F. Celiac disease: how complicated can it get? Immunogenetics 2010;62:641-51. [ Links ]
66. Green ph, Jabri B. Coeliac disease. Lancet 2003;362:383-91. [ Links ]
67. Pinier M, Fuhrmann G, Verdu EF, Leroux JC. Prevention measures and exploratory pharmacological treatments of celiac disease. Am J Gastroenterol 2010;105:2551-61; quiz 62. [ Links ]
Mariana Cunha
Serviço Anatomia patológica, Centro Hospitalar S.João
Alameda prof. Hernâni monteiro 4200 – 319 Porto
Email: mariana.mc87@gmail.com

