










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.27 no.3 Porto jun. 2013
ARTIGO DE REVISÃO
Neoplasias primárias do fígado em idade pediátrica – da patologia à clínica
Primary liver tumors in pediatric population -from pathology to clinic
Ana Rodrigues1, Helena Baldaia2, Fátima Carneiro2,3
1 Faculdade de Medicina da Universidade do Porto
2 Serviço de Anatomia Patológica, Centro Hospitalar de São João departamento de Patologia e Oncologia, Faculdade de Medicina da Universidade do Porto
3 Instituto de Patologia e imunologia Molecular da Universidade do Porto
RESUMO
As neoplasias primárias do fígado em idade pediátrica englobam um espetro complexo e amplo de tumores epiteliais, mesenquimatosos e vasculares, com comportamentos biológicos distintos (desde achados incidentais benignos a lesões malignas). Por serem entidades raras e com uma apresentação clínica inicialmente inespecífica, exigem frequentemente um elevado nível de suspeição para permitir um diagnóstico atempado. A epidemiologia, os achados clínicos e analíticos, os aspetos imagiológicos e sobretudo as características morfológicas e moleculares das lesões permitem atualmente um diagnóstico mais preciso destas entidades, com implicações prognósticas e terapêuticas. A monografia apresentada tem como objetivo a revisão da epidemiologia, da etiopatogénese, da histopatologia e das alterações moleculares das neoplasias primárias do fígado mais frequentes em idade pediátrica.
Palavras-chave: Fígado, patologia, tumor hepático pediátrico, tumor epitelial, tumor mesenquimatoso, tumor vascular
ABSTRACT
Primary liver tumors in children and adolescents include a wide and complex spectrum of epithelial, mesenchymal and vascular neoplasms, ranging from benign, incidentally discovered lesions to malignant entities. A high level of suspicion is usually required for an early diagnosis of these tumors, due to their rarity and unspecific clinical presentation. The current knowledge on the clinical, analytical and imaging features as well as on the (molecular) pathology profile contributes for an earlier diagnosis, with prognostic and therapeutic implications. This paper reviews the epidemiology, etiopathogenesis, histopathology and molecular features of the most common primary liver tumors in pediatric population.
Key-words: Liver, pathology, pediatric liver tumor, epithelial neoplasms, mesenchymal neoplasms, vascular neoplasms
INTRODUÇÃO
As neoplasias primárias do fígado são entidades raras em idade pediátrica, embora o achado radiográfico de uma lesão hepática focal não seja invulgar nesta faixa etária,1 implicando o diagnóstico diferencial com lesões inflamatórias, císticas ou metastáticas.2,3 de facto, na suspeita de neoplasia hepática, as metástases (sobretudo de neuroblastoma e tumor de Wilms) estão implicadas na maioria dos casos.1,3,4
O progresso nos cuidados de saúde materno-infantil tem-se acompanhado de um discreto aumento da incidência de tumores hepáticos em idade pediátrica, refletindo a melhoria da sobrevida dos prematuros e o investimento em métodos de rastreio imagiológico.3,5 Por outro lado, a introdução da vacinação contra o vírus da hepatite B em países do sudeste Asiático tem sido associada a uma alteração das frequências relativas dos diferentes tumores hepáticos, com uma redução marcada da incidência de hepatocarcinoma (HC) em crianças.5-7
Os tumores primários do fígado representam 0,32% de todos os tumores pediátricos e dois terços são malignos.2,4 nos EUA, cerca de 1% dos tumores malignos que ocorrem antes dos 20 anos têm origem primária no fígado.7 nos países ocidentais, o hepatoblastoma (HB) lidera com 43%, seguido de HC (23%), tumores vasculares (14%), sarcoma (embrionário) indiferenciado (SEI) (7%), hamartoma mesenquimatoso (HM) (6%), adenoma hepatocelular (AHC) (2%) ehiperplasia nodular focal (HNF) (2%).2 Nas neoplasias hepáticas com apresentação fetal ou neonatal, os tumores vasculares constituem 60% dos casos, seguidos do HM (23%) e do HB (16%).8
Apesar do amplo espetro de tumores epiteliais, mesenquimatosos e vasculares, a apresentação clínica é semelhante, sobretudo em crianças pequenas, com distensão abdominal não dolorosa e sintomas específicos de aparecimento tardio.2,4 As características morfológicas das lesões, aliadas à informação clínica e imagiológica possibilitam o diagnóstico e a apreciação do prognóstico.9
O trabalho de revisão bibliográfica apresentado tem como objetivo a análise das neoplasias hepáticas primárias mais frequentes em idade pediátrica. Para cada uma das entidades os autores procuraram rever os aspetos relacionados com o perfil epidemiológico, etiopatogénese e histopatologia, relacionando, quando possível, o prognóstico com as características morfológicas e/ou moleculares das lesões.
MÉTODOS
O estudo consistiu numa revisão da literatura publicada, acessível através da base de dados Pubmed/ medline. A pesquisa inicial foi limitada a artigos originais ou de revisão, publicados em português ou inglês, desde 1 de janeiro de 2000, e a estudos realizados em humanos. os termos utilizados (pediatric liver tumor) permitiram a identificação de 1068 artigos indexados, tendo sido selecionados 397 com base no título. Posteriormente procedeu-se à leitura dos resumos, com vista a incluir apenas os estudos enquadráveis no âmbito definido. Foram excluídos os artigos que se reportavam a uma população com idade superior a 18 anos, que não se relacionavam comos objetivos propostos e aqueles cujo resumo e/ou texto integral não estavam disponíveis. No trabalho de revisão foram ainda considerados artigos que, não tendo derivado diretamente da pesquisa inicial, constavam da lista de referências de estudos selecionados e o capítulo liver tumors of childhood do livro Practical hepatic pathology: a diagnostic approach.
A organização do artigo em três secções (tumores epiteliais, mesenquimatosos e vasculares) seguiu a estrutura mais frequentemente utilizada na literatura.
TUMORES EPITELIAIS
Hiperplasia nodular Focal
A HNF é uma lesão benigna, resultante da proliferação policlonal de hepatócitos, células de Kupffer, estruturas vasculares e ductos biliares.1,4 É uma entidade rara em idade pediátrica, mais frequente no género feminino.1,10,11 O diagnóstico é efetuado em cerca de 8% dos casos antes dos 15 anos.6 os níveis de a-fetoproteína (AFP) são normais.11
Embora a etiopatogénese desta lesão seja ainda controversa, admite-se que possa resultar de uma anomalia vascular congénita ou adquirida, condicionando distúrbios circulatórios focais e acompanhando-se, sucessivamente, de trombose vascular, recanalização e reperfusão, com resposta de hiperplasia hepatocelular.12,13 Na literatura encontram-se descritos casos de HNF em crianças submetidas a quimioterapia por neoplasias sólidas malignas, tendo-se postulado que alguns dos fármacos utilizados possam induzir lesões vasculares, nomeadamente, doença venoclusiva.10,12 Ocasionalmente verifica-se coexistência ou conversão de hemangioma infantil (HI) em HNF, sugerindo que ambas as lesões possam ter origem em anomalias vasculares.14 na literatura encontram-se reportadas alterações moleculares de significado pouco esclarecido, afetando os genes da angiopoietina (ANGPT1 e ANGPT2)(com aumento do rácio ANGPT1/ANGPT2 e diminuição da expressão de ANGPT2)15 e a via da ß-catenina (com ativação).16
A HNF é frequentemente solitária (sendo multifocal em cerca de um terço dos casos),1,10 bem limitada, sem cápsula.4,17,18 hemorragia ou necrose são achados raros.1,13
Na histologia descrevem-se septos fibrosos que delimitam nódulos coalescentes de hepatócitos hiperplasiados, constituindo uma cicatriz central em forma de estrela.4,18 Pode igualmente observar-se proliferação colangiolar, com formação de dúctulos.1 A cicatriz central é composta por estroma mixomatoso com vasos distróficos, igualmente presentes nos septos fibrosos que aí se originam.1,13 No estudo imunohistoquímico observa-se expressão de CD34 nas estruturas vasculares e de citoqueratina 18 e marcadores hepatocitários nos elementos celulares hiperplasiados.11 A sobre-expressão da sintetase da glutamina origina um padrão típico em mapa geográfico.19
A HNF apresenta um crescimento lento e raramente cursa com complicações.10,19 Por se desconhecer potencial maligno, o tratamento é geralmente conservador,1,4,16 recorrendo-se à biópsia no caso de evolução clínica e/ou imagiológica atípica.10,17 Na presença de sintomas, a ressecção cirúrgica ou a ablação podem estar indicadas.1,4,19
Adenoma Hepatocelular
O AHC é uma neoplasia benigna, rara em idade pediátrica. Associa-se frequentemente ao uso de esteroides (contracetivos orais, androgénios) e a distúrbios metabólicos, particularmente glicogenoses dos tipos I/III, galactosemia e diabetes mellitus familiar.1,20-22 A distribuição etária é tipicamente bimodal em crianças sem doença metabólica ou fatores de risco, com o primeiro pico nos primeiros dois anos de vida e o segundo na puberdade; no contexto de doença metabólica surge sobretudo em crianças com mais de cinco anos.6 Os níveis de AFP são normais.1,23-25
A neoplasia é solitária em cerca de 70-80% dos casos, sendo mais frequentemente múltipla no contexto de doença metabólica ou terapia androgénica.1,23 Quando estão presentes mais de dez adenomas, na ausência de doença hepática subjacente ou fator de risco conhecido, aplica-se o termo adenomatose, associando-se a maior risco de hemorragia e transformação maligna.24
Na Tabela 1 resumem-se as características do AHC.
Hepatoblastoma
O HB é a neoplasia maligna primária do fígado mais frequente em crianças, representando 1% de todos os tumores pediátricos.26 Surge caracteristicamente nos dois primeiros anos de vida27 e, na maioria dos casos (80-90%), antes dos cinco,6,7 com um ligeiro predomínio no género masculino.7 O diagnóstico pode ser realizado no período pré-natal.4,26 Em cerca de 90% dos doentes verifica-se elevação dos níveis de AFP.4,28 Valores séricos normais ou diminuídos associam-se a um curso clínico agressivo (com extensão local ou doença metastática) e a subtipos de HB de alto risco (HB indiferenciado de pequenas células).28-30
Nas últimas décadas tem-se assistido a um ligeiro aumento da incidência de HB no mundo ocidental.26 Embora tal se atribua à melhoria da sobrevida de recém-nascidos com baixo/muito baixo peso, o mecanismo etiopatogénico não se encontra bem esclarecido.31 Uma das hipóteses é a de que a exposição dos hepatoblastos a agentes endógenos/exógenos (normalmente eliminados pela placenta) facilita modificações do genoma;3 outras propõem que os agentes terapêuticos empregues na manutenção dos recém-nascidos podem ter efeito carcinogénico31 ou que a presença de anomalias genéticas pode explicar simultaneamente a imaturidade e o desenvolvimento neoplásico.32
O HB associa-se à síndrome de Beckwith-Wiedemann (SBW), à Polipose Adenomatosa familiar (PAF), à Síndrome de Gardner, à glicogenose tipo 1A, à Síndrome de Li-Fraumeni e à trissomia18.5,27,33
A neoplasia é geralmente bem limitada, solitária, com contornos lobulados,34 descrevendo-se áreas de hemorragia e necrose27 e, ocasionalmente, septos27 e calcificações.29 A maioria das lesões surge no lobo direito.29
Classicamente, o HB é classificado em dois grandes grupos: tipo epitelial [subtipos: fetal (Figura 1); embrionário; macrotrabecular; indiferenciado de pequenas células] (Tabela 2) e tipo misto (com/ sem características teratoides).6,29,35,36 O HB misto é constituído por um componente epitelial (mais frequentemente fetal/embrionário) e um componente mesenquimatoso. Na variante teratoide (correspondente a cerca de 3% dos HB mistos) identificam-se células neoplásicas de origem neuroectodérmica ou endodérmica.6
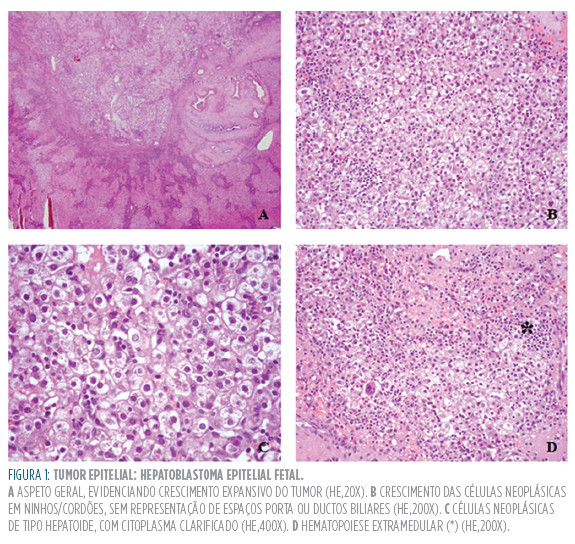
No HB submetido a quimioterapia ocorre diminuição da massa tumoral e aumento da área de hemorragia e necrose; ocasionalmente documenta-se apenas osteoide ou focos de epitélio escamoso.6 nas lesões que regridem, a presença de necrose e fibrose/ esclerose é comum.6,37
Do ponto de vista citogenético, os HB exibem anomalias cromossómicas (envolvendo 1q, 2q, 4q, 8, 11q, 17q, 20), ainda sem significado clínico bem definido.38 os ganhos cromossómicos são mais comuns do que as perdas.39 Ganhos nas regiões 2q, 8q ou 20 podem associar-se a um curso clínico agressivo.40 Translocações não equilibradas envolvendo a região terminal do 4q e as trissomias 2, 8 e 20 podem ser identificadas isoladamente, sugerindo tratar-se de eventos precoces na oncogénese.39,41,42 Encontram-se descritas perdas de heterozigotia nas regiões 1p,1qe11p,bem como a sobre-expressão do gene MET.38,43
Os HB podem apresentar alterações na via de sinalização Wnt/ß-catenina.38 As mutações missense ou deleções intersticiais do gene da ß-catenina são as alterações moleculares mais frequentes no HB esporádico, condicionando acumulação citoplasmática e/ou nuclear da proteína.38,44 Com base na elevada frequência de mutações e na ausência de associação com o tipo histológico, postula-se que as alterações do gene da ß-catenina representem um evento precoce na oncogénese.44,45 Encontram-se descritas igualmente alterações de outros elementos da via Wnt/ß-catenina, nomeadamente, dos genes das axinas 1 e 2 e APC.38 Na PAF, mutações no gene APC condicionam aumento da expressão da ß-catenina e explicam a associação do HB a esta entidade,29 embora as mutações ocorram fora da região associada à PAF clássica.46 Em pacientes com SBW, o elevado risco de HB relaciona-se com alterações no imprinting dos genes localizados na região 11p15 (hipermetilação e silenciamento do locus IGF-II/H19).6 Outros genes podem estar alterados no HB, nomeadamente: CCND1;47 TP53;39 GLI1, PTCH1 e BCL2;48 PLK1;6 FOXG1;49 p27;50 p16.51
Os três fatores prognósticos mais relevantes no HB são o estadio clínico, os níveis de AFP e os tipos histológicos (Tabela 1).26,28,52 A sobrevida global ronda os 70%53 e a ressecção cirúrgica associada a quimioterapia é o tratamento de eleição.28 A extensão tumoral, a presença de metástases e a multifocalidade afetam negativamente o prognóstico.26
Para além dos tipos de HB apresentados, foram reportadas variantes raras, nomeadamente a papilar e mixoide6 e o HB colangioblástico.54 no grupo de neoplasias relacionadas com o HB incluem-se ainda os tumores da placa ductal54 e o tumor de células de transição do fígado.6,55
Tumor epitelial-estromal em ninhos (calcificantes)
Trata-se de uma entidade rara, caracterizada recentemente e reportada quase exclusivamente na idade pediátrica, com potencial maligno reduzido e comportamento indolente.56-58 os níveis de AFP são normais.56,57
O tumor apresenta-se sob a forma de massa solitária, bem limitada, sem cápsula.56,57 na histologia documenta-se um padrão organoide com ninhos celulares constituídos por células epitelioides e/ou fusiformes, envolvidos por miofibroblastos.56,57O estroma é fibroso e denso, contendo estruturas vasculares.56 ocasionalmente, observa-se proliferação biliar que envolve focalmente os ninhos celulares.56,58 A ossificação, os focos de osteoide e as calcificações psamomatosas são achados ocasionais.56-58 Os ninhos celulares expressam EMA e citoqueratinas, nas células epitelioides, e vimentina e WT-1, nas células fusiformes.56,57 Os miofibroblastos apresentam imunorreatividade para vimentina e actina do músculo liso e no componente biliar observa-se expressão de citoqueratinas 7 e 19.56,58 A etiopatogénese deste tumor não está bem esclarecida, tendo-se postulado a sua origem num precursor mesenquimatoso.56,57
Hepatocarcinoma
O HC é a segunda neoplasia maligna do fígado mais frequente em idade pediátrica, ultrapassando o HB em regiões de alta incidência de infeção pelo vírus da hepatite B.26,27,59 Afeta usualmente crianças mais velhas e adolescentes, com predomínio do género masculino.27,59,60 Os níveis de AFP encontram-se tipicamente elevados.59
Em idade pediátrica, a maioria dos HC surge de novo 60,61 e apenas 20-35% ocorrem em contexto de doença hepática crónica ou cirrose (6). Os fatores de risco para estas condições incluem doenças metabólicas (glicogenoses, deficiência de a1-antitripsina, doença de Wilson, tirosinemia hereditária), atrésia biliar, síndrome de Alagille, síndromes colestáticos intra-hepáticos familiares e infeção pelos vírus da hepatite B/c.5,27,59,62 em áreas de elevada incidência, a infeção pelo vírus da hepatite B constitui a causa mais comum de HC em crianças e adolescentes.60
O HC pode apresentar-se como uma lesão solitária, multinodular ou, mais raramente, com crescimento difuso.27 O envolvimento de ambos os lobos é frequente.61
A histologia do HC pediátrico é idêntica à do adulto.6 o tumor pode ser envolvido por cápsula fibrosa27 e apresentar padrões trabeculares, acinares ou compactos/sólidos (em formas puras ou em associação).6 A invasão vascular, a hemorragia e a necrose são achados frequentes.27,34 A neoplasia é constituída por células semelhantes a hepatócitos, com diversos graus de diferenciação, citoplasma abundante e eosinofílico e núcleos com um ou mais nucléolos proeminentes.34 Raramente, o tumor pode ser composto por células claras.61,63 As células neoplásicas podem acumular ferro, cobre ou lípidos e conter corpos de Mallory ou glóbulos hialinos PAS positivos.6 As células neoplásicas expressam frequentemente citoqueratinas7, 8, 18, 20 e o endotélio vascular apresenta imunorreatividade para CD34.6
As alterações moleculares do HC pediátrico são diferentes das observadas no HC do adulto, caracterizando-se por mutação do MET, perda de heterozigotia na região13q e diminuição da expressão de ciclina D1.60 As alterações estruturais cromossómicas são mais frequentes no HC do que no HB, estando descritas múltiplas anomalias, nomeadamente, ganhos das regiões 1q, 8q e 17q e perdas de 4q, 8p, 13q, 16q e 17p. Alterações da via de sinalização Wnt/ßcatenina também se encontram descritas no HC, sendo mais frequentes as mutações missense do gene da ß-catenina e raras as mutações nos genes que codificam as axinas.38
O prognóstico do HC em idade pediátrica é reservado, com sobrevida a longo prazo de 10-30%,59 influenciada pela multifocalidade, presença de invasão vascular e estadio no momento do diagnóstico.60,61 Apesar da relativa sensibilidade à quimioterapia, os protocolos são pouco eficazes e a ressecção cirúrgica radical ou o transplante hepático constituem as melhores opções terapêuticas.27,59,60 Após ressecção cirúrgica, a sobrevida é superior à verificada nos adultos, por ser menor o risco de falência hepática ou de recorrência.60
Carcinoma fibrolamelar
O carcinoma fibrolamelar (CFL) é uma neoplasia maligna da família dos HC, ocorrendo mais comummente em adolescentes e jovens (entre os 1035 anos), na ausência de doença hepática crónica ou cirrose.27,64-67 A etiologia desta entidade é ainda desconhecida,66 não estando associada a hepatite vírica, álcool ou outros fatores de risco tradicionalmente associados a HC.64,67 Os níveis de AFP são normais.64,66-68
O CFL é geralmente solitário, bem limitado,27,66 atingindo o lobo esquerdo em cerca de dois terços dos casos.6 Na histologia documentam-se células epiteliais poligonais, bem diferenciadas, de grandes dimensões, com citoplasma granular e eosinofílico e nucléolos proeminentes.27,64,66,69 As células neoplásicas, organizadas em cordões e trabéculas, são envolvidas por estroma fibroso abundante, de estrutura lamelar, com cicatriz central, onde se podem observar calcificações.6,64,66 Em cerca de 30-50% das células neoplásicas documentam-se corpos pálidos – inclusões eosinofílicas citoplasmáticas.6 Ocasionalmente descrevem-se glóbulos hialinos PAS positivos70 e granulomas epitelioides.64 A colestase é um achado frequente e explica a deposição de cobre descrita em alguns casos.64 Foram reportadas duas variantes: uma de células claras e outra com formações pseudoglandulares e produção de mucina.64,66
As células neoplásicas expressam marcadores hepatocitários e citoqueratina 7 e, de forma variável, EMA, fibrinogénio (nos corpos pálidos), ferritina e a1-antitripsina.6,66,69
A patogenia molecular é pouco conhecida, considerando-se o CFL uma entidade estável do ponto de vista cromossómico, sem mutações dos genes habitualmente envolvidos na etiopatogénese do HC.6,64,66 As anomalias citogenéticas mais frequentes envolvem ganhos nas regiões 1q e 8q e perdas na 18q.69 As alterações do gene tP53 são raras e não há evidência de ativação da via Wnt/ßcatenina; na maioria dos casos documenta-se sobre-expressão do EGFR.64
Embora se associasse o CFL a um prognóstico melhor do que o do HC do adulto, tal parece dever-se à ausência de cirrose, à idade jovem no momento do diagnóstico e ao crescimento expansivo do tumor,64-67 não se tendo encontrado diferenças significativas na resposta ao tratamento das duas entidades.68 em geral, o prognóstico do CFL é melhor do que o do HC tipo adulto em contexto de cirrose, mas idêntico ao do HC que surge em fígados não cirróticos.64 A ausência de invasão vascular é também um fator de bom prognóstico.65,67,68
TUMORES MESENQUIMATOSOS
Hamartoma mesenquimatoso
O HM é a segunda neoplasia hepática benigna mais comum na infância, apresentando-se frequentemente sob a forma de massa multicística.71 cerca de 85% destes tumores ocorrem em crianças com menos de três anos34,71 e menos de 5% são diagnosticados depois dos cinco.71
A neoplasia apresenta-se individualizada, apesar da ausência de cápsula,6,34 estando descrita multifocalidade, com lesões satélites à periferia do tumor principal.72 Em 85% dos casos documentam-se espaços císticos, sem comunicação direta com o trato biliar, eventualmente ausentes em doentes muito jovens.6,34,71 áreas de necrose, calcificação ou hemorragia são raras.34
Na histologia, o tumor é constituído por tecido conjuntivo laxo, ductos biliares de pequenas dimensões, cordões hepatocitários e cistos.6 O componente mesenquimatoso é caracteristicamente mixoide, hipocelular, formando bainhas em torno de ductos biliares e vasos tortuosos.6,71 A presença de hepatócitos suporta o achado ocasional de níveis moderadamente elevados de AFP.1,73,74 O estroma contém ainda um número variável de espaços císticos: os cistos de menores dimensões encontram-se revestidos de epitélio cuboide, com expressão de citoqueratinas 7, 8 e 19, suportando a hipótese de origem biliar; os cistos de maiores dimensões não apresentam revestimento epitelial e derivam provavelmente de regiões de mesênquima com degenerescência.71 Em mais de 85% dos casos observa-se hematopoiese extramedular.6
Na literatura estão descritos casos de coexistência de HM com hi75 ou com cisto solitário congénito.76 A designação de hamartoma mioide foi proposta para os casos com diferenciação muscular no componente mesenquimatoso.77
A patogénese do HM não está bem esclarecida, tendo sido propostas quatro teorias: i) perturbação do desenvolvimento (malformação da placa ductal);6,71 ii) anomalia da vascularização;71 iii) estímulo tóxico (o componente mesenquimatoso do tumor expressa vimentina, actina do músculo liso e desmina, aspetos sugestivos da ativação de células de Ito);71,78 iv) desenvolvimento neoplásico (sustentada pelo achados ocasionais de aneuploidia ou translocações equilibradas envolvendo a região 19q13.4).1,6,71,73,79
Embora o HM seja considerado um tumor benigno, há casos descritos de associação com SEI,71,79,80 sugerindo uma possível origem comum em células mesenquimatosas indiferenciadas.34 Alguns aspetos morfológicos semelhantes e alterações citogenéticas da região 19q13 suportam esta hipótese.71,79
O prognóstico é favorável na maioria dos casos.6 Apesar de estar documentada regressão espontânea,81 a associação entre HM e SEI implica uma ponderação adequada do risco de malignidade e seguimento a longo prazo quando se opta por uma abordagem conservadora.71
Sarcoma (embrionário) indiferenciado
O SEI é uma neoplasia maligna de origem mesenquimatosa, ocorrendo caracteristicamente entre os seis e os dez anos,6,82 raramente reportada em idades mais jovens e adultos.82-84 É a quarta neoplasia hepática mais frequente em idade pediátrica,2,82 perfazendo 9-15% das neoplasias malignas desse órgão.34,82 os níveis de AFP são normais.27,84,85
O tumor é geralmente solitário e sólido, podendo apresentar uma ou mais lesões císticas centrais, hemorragia e necrose, até 80% da superfície de corte (Figuras 2A-B).6,82 O SEI é bem limitado, frequentemente com uma pseudocápsula fibrosa.27,82,86
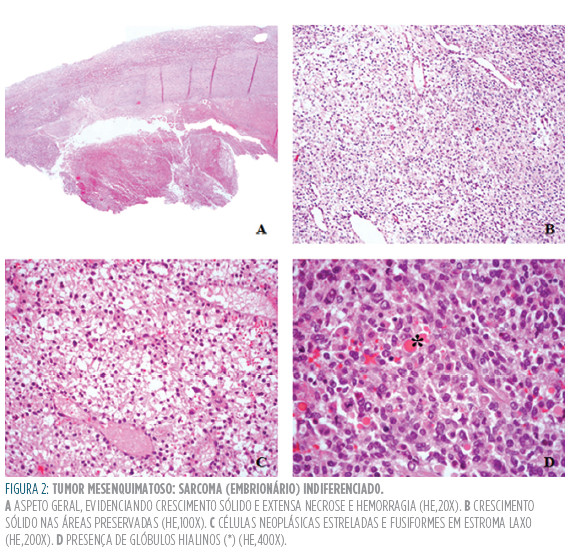
Na histologia documenta-se tecido mixoide abundante com células neoplásicas fusiformes e estreladas, sem evidência de estriação, e elementos polimórficos (Figura 2C).6,27,86,87 O tumor pode conter ductos biliares (ou perfis de estruturas similares) na periferia do tumor6,27 e/ou ninhos de hepatócitos sem características de malignidade no interior da lesão, sugerindo tratar-se de estruturas do parênquima não neoplásico encarceradas na frente de invasão.6,27,82 focos de hematopoiese extramedular são comuns.82,87O citoplasma das células pleomórficas contém glóbulos hialinos PAS positivos (Figura 2D)6,82,86 que podem estar igualmente presentes na matriz extracelular.87 As mitoses são comuns27,86 e o achado de células multinucleadas bizarras é frequente.82,84,86,87
As células neoplásicas expressam vimentina e cd38 e, mais raramente, desmina,86 a1-antitripsina,84,88,89 a1-quimotripsina89 e citoqueratinas.90
O SEI apresenta frequentemente rearranjos da bandacromossómica19q13.4,91 incluindo a translocação t(11;19)(q13;q13.4)6,79 ou a t(11;19)(q11;q13.4).92 Sowery et al91 caracterizaram ainda ganhos nas regiões 1q, 5p, 6q, 8p, 12q e perdas nos braços 9p, 11p e cromossoma 14, tendo sido sugerido que algumas das alterações citogenéticas descritas se deveriam a fenómenos aleatórios e outras seriam efetivamente responsáveis pelo fenótipo maligno.
No passado, o prognóstico do SEI era considerado reservado, com sobrevida média de 12 meses, mesmo com ressecção cirúrgica.82 As terapêuticas multimodais permitiram melhorar a sobrevida dos doentes, mesmo naqueles com tumores inicialmente não ressecáveis ou resistentes à quimioterapia.85,93 Atualmente, na ausência de sinais de recidiva ou metástase dois anos após a cirurgia, a sobrevida é longa.94
Rabdomiossarcoma
O rabdomiossarcoma (RMS) é a neoplasia maligna da árvore biliar mais comum em crianças, representando cerca de 1% de todos os RMS pediátricos6,27,34 e ocorrendo principalmente entre os dois e os quatro anos.34,95 os níveis de AFP são normais.27
O tumor envolve mais frequentemente os ductos extra-hepáticos mas pode ter crescimento intrahepático ab initio ou por disseminação local.27,82 o crescimento tumoral inicia-se nos ductos biliares de médio/grande calibre, constituindo-se massas que se insinuam na mucosa do ducto e que formam projeções em cacho de uva (padrão de crescimento botrioide).6,82
Na histologia descrevem-se dois tipos de RMS: embrionário (mais comum) ou botrioide.4 no tipo embrionário documenta-se tecido laxo (mixoide), com células neoplásicas pequenas, frequentemente estreladas.6,34 Os rabdomioblastos apresentam núcleos pequenos e densos, com baixo índice mitótico,6,27 e estriação citoplasmática ocasional.34,82,90 no tipo botrioide, as massas polipoides encontram-se revestidas por epitélio biliar cuboide,27,82 subjacente ao qual se observa uma banda compacta de elevada celularidade (cambium layer).6,27 As células neoplásicas expressam desmina, miogenina e Myod.6,27,90
Embora o crescimento tumoral ocorra ao longo dos ductos biliares em ambos os lobos,6,27 a intervenção precoce com ressecção cirúrgica e tratamento multimodal tem contribuído para um melhor prognóstico,27 com sobrevida de 60-70% a longo prazo.4
Tumor rabdoide Maligno do Fígado
O TRM do fígado é uma entidade rara, com comportamento clínico agressivo,96-98 ocorrendo em cerca de 89% dos casos em crianças com menos de dois anos.97
O tumor forma massas lobuladas com necrose e hemorragia.6,99,100 Na histologia exibe um crescimento difuso e desorganizado, sendo constituído por células poligonais de pequena/média dimensão, uniformes, não coesas, com citoplasma eosinófilo, no seio deestromafibromixoide.6,97-100 As células apresentam núcleo pleomórfico, vesicular, em localização excêntrica e um corpo esferoide e o sinofílico paranuclear.6,97-99 Alguns TRM são constituídos exclusivamente por células de pequenas dimensões, dificultando
O diagnóstico diferencial com HB indiferenciado de pequenas células.6 A organização das células neoplásicas em ninhos, estruturas trabeculares ou pseudotúbulos é rara.101 ocasionalmente observam-se células pleomórficas e multinucleadas.101
Nas células tumorais observa-se expressão de vimentina, cd99 e EMA mas não de marcadores mioides.96,98,99,101,102 os corpos paranucleares expressam vimentina e, ocasionalmente, citoqueratinas, a traduzir a natureza dos filamentos intermediários que os constituem.6,96-98,101 As células neoplásicas são negativas para ini1.6,96,98,101 Alguns TRM do fígado apresentam deleção da banda cromossómica 22q11, correspondente ao locus do gene SMARCB1/ INI1.6,98,102 Esta característica auxilia no diagnóstico diferencial com HB indiferenciado de pequenas células, uma vez que ambos se podem associar a níveis reduzidos de AFP.30,98
O TRM do fígado é um tumor muito agressivo, com prognóstico reservado e mortalidade próxima dos 90%.6,97,98
TUMORES VASCULARES
Hemangioma infantil
O HI (ou hemangioendotelioma infantil) é o tumor hepático benigno mais frequente na infância,1 sendo diagnosticado em 80% dos casos nos primeiros seis meses de vida,6 mais frequentemente no género feminino.1 A história natural da lesão caracteriza-se pelo crescimento pós-natal rápido (durante 9-12 meses), seguido de regressão espontânea (durante 5-7 anos), com substituição da lesão inicial por tecido fibroadiposo.103 os níveis de AFP raramente excedemos valores de referência;104 quando tal sucede, a elevação é inferior à verificada no HB.105
Classicamente definiam-se dois tipos de HI: tipo 1 (capilares ou cavernosos) e tipo 2 (compactos e hipercelulares).6,106 Os HI tipo 2 são atualmente considerados variantes do angiossarcoma pediátrico (ASP); no caso de lesões múltiplas ou difusas deve-se suspeitar desta última entidade.6 Mo et al107 descreveram, pela primeira vez, a expressão de GLUT1 em HI hepáticos, à semelhança do observado previamente em hemangiomas cutâneos. A reatividade foi ainda observada em lesões de HI em regressão e, focalmente, em angiossarcomas com origem em HI prévio.107 na classificação proposta por Christison-Lagay et al103 consideraram-se três categorias distintas de HI: i) focais (geralmente assintomáticos, sem expressão de GLUT1); ii) multifocais (com expressão de GLUT1 e regressão espontânea; associação a hemangiomas cutâneos); iii) difusos (envolvimento global do parênquima hepático; associação a sintomatologia exuberante e hipotiroidismo). Foi sugerido que a associação ao hipotiroidismo resulta da atividade da desiodinase da iodotironina tipo 3 no tecido tumoral, condicionando degradação das hormonas tiroideias.108 A remissão espontânea do hipotireoidismo resulta normalmente da regressão do HI.109 Com base nesta associação e na expressão de GLUT1, alguns autores sugeriram a possibilidade do HI ter origem em angioblastos ou células endoteliais de origem placentar.107,110
Na histologia, o HI caracteriza-se pela presença de canais vasculares, no seio de estroma fibroso, revestidos por uma camada única de células endoteliais, com expressão de CD34 (Figura 3).1,6 As mitoses são raras e a hematopoiese luminal deteta-se em cerca de dois terços dos tumores.6 Em fase de regressão, é comum a descrição de espaços vasculares cavernosos na área central, delimitados por uma única camada endotelial e com evidência de trombose, fibrose e/ou calcificação.1,107
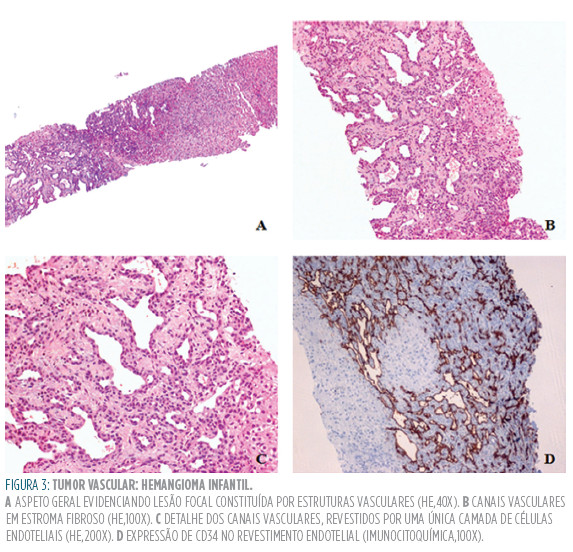
O prognóstico do HI é geralmente excelente, sobretudo no caso de regressão espontânea.1,104 Os casos sintomáticos e não tratados de forma atempada podem apresentar uma evolução clínica menos favorável, com insuficiência cardíaca, compromisso respiratório e síndrome do compartimento abdominal, entre outras complicações.1,6
Hemangioendotelioma epitelioide
O hemangioendotelioma epitelioide (HE) atinge muito raramente o fígado em idade pediátrica,111,112 afetando adolescentes dos 12-14 anos.6 Trata-se de uma entidade de crescimento lento, com potencial maligno intermédio (entre HI e ASP).111,112 Os níveis de AFP são normais.111,112 no HE são reconhecidos dois padrões que normalmente se sucedem: i) multifocal, caracterizado por múltiplos nódulos periféricos que aumentam de dimensões e coalescem; ii) difuso, típico de fases mais avançadas. As massas tumorais podem provocar retração da cápsula hepática por reação fibrótica.27 A neoplasia é constituída por células epitelioides (com morfologia idêntica a células epiteliais mas com diferenciação vascular) e células fusiformes. o centro do tumor é relativamente hipocelular e avascular, composto primariamente por estroma (mixoide ou hialinizado).27,112 As células neoplásicas expressam antigénio associado ao fator VIII e marcadores endoteliais (cd31, CD34).112 Nos casos descritos em idade pediátrica, o HE tem um curso clínico mais agressivo do que nos adultos.111
Angiossarcoma Pediátrico
O ASP é uma neoplasia maligna e agressiva, muito rara em idade pediátrica,27,33 diagnosticada mais frequentemente em crianças pequenas (40-45 meses de idade).6,27,113 Não se encontra descrita associação aos fatores de risco ambientais descritos para o angiossarcoma do adulto (torotraste, arsénio e cloreto de vinilo).27,113 Raramente, o ASP pode ser precedido por um HI.33,114,115 Na suspeita de uma lesão de HI em crianças com mais de um ano de idade deve ser instituída vigilância pelo risco de transformação maligna.113 Quando se verifica crescimento rápido do tumor, aparecimento de metástases, ausência de resposta ao tratamento ou recorrência de uma lesão de HI, deve ser considerada a hipótese de ASP, mesmo na ausência de características histológicas de malignidade.113,116
A neoplasia é geralmente multifocal, com envolvimento de ambos os lobos 27,104,113 por massas multinodulares, com necrose e hemorragia.6,27,113 Na histologia, as células neoplásicas apresentam crescimento compacto e não formam canais vasculares.6 As células podem ser pleomórficas ou fusiformes,27 as últimas com glóbulos hialinos PAS positivos (aspeto kaposiforme).6,117 Ocasionalmente os canais vasculares podem apresentar morfologia irregular e serem delimitados por múltiplas camadas de células neoplásicas pleomórficas, com configuração papilar em áreas focais.113,117 As células neoplásicas expressam CD31 e CD34.6,113,117 As células fusiformes são negativas para o antigénio associado ao fator VIII6,113 mas expressam a1antitripsina e a1-quimiotripsina.113
O prognóstico do ASP é muito reservado, com uma sobrevida média de dez meses a dois anos, apesar da abordagem terapêutica multimodal com ressecção cirúrgica,quimioterapia e radioterapia.4,104,113
CONCLUSÃO
O espetro diversificado de tumores hepáticos em crianças e adolescentes é muito diferente do observado em adultos e, salvo algumas exceções, exclusivo dessa faixa etária. Embora sejam entidades raras, a sua relevância clínica advém do facto de frequentemente não ser possível o diagnóstico atempado. A implementação de programas de rastreio e vigilância em situações clínicas de alto risco deve ser ponderada, uma vez que pode permitir uma intervenção precoce, potencialmente curativa.
Nos últimos anos, novas entidades foram descritas, com significado biológico pouco esclarecido. Dada a complexidade e raridade de alguns destes diagnósticos, urge a cooperação de grupos multidisciplinares, com a criação de bases de dados que permitam o registo de um maior número de casos e a acumulação de experiência que fundamente a tomada de decisões baseada na evidência.
Com base nos achados morfológicos e moleculares descritos na literatura, postula-se que algumas das lesões apresentadas possam representar processos que ocorrem na ontogénese hepática normal. Contudo, a implicação de tais hipóteses na classificação dos tumores é ainda incerta. Acredita-se que a melhor compreensão do comportamento biológico das neoplasias hepáticas e das alterações moleculares subjacentes permitirá o desenvolvimento de terapêuticas dirigidas e mais eficazes do que as empregues na atualidade.
REFERÊNCIAS
1. Chung Em, Cube R, Lewis RB, Conran Rm. from the archives of the AfiP: pediatric liver masses: radiologic-pathologic correlation part 1. Benign tumors. Radiographics 2010;30:801-26. [ Links ]
2. Von Schweinitz D. Management of liver tumors in childhood. Semin Pediatr surg 2006;15:17-24. [ Links ]
3. Hadzic N, Finegold MJ. Liver neoplasia in children. Clin liver dis 2011;15:443-62. [ Links ]
4. Meyers RL. Tumors of the liver in children. Surg oncol 2007;16:195-203. [ Links ]
5. Litten JB, Tomlinson GE. Liver tumors in children. Oncologist 2008;13:812-20. [ Links ]
6. Zimmermann A. Liver tumors of childhood. in: Saxena R, editor. Practical hepatic pathology: a diagnostic approach. 1st ed. Philadelphia: saunders;2011. p. 521-46. [ Links ]
7. Darbari A, Sabin Km, Shapiro CN, Schwarz KB. Epidemiology of primary hepatic malignancies in U.S. children. Hepatology 2003;38:560-6. [ Links ]
8. Isaacs H. fetal and neonatal hepatic tumors. J Pediatr surg 2007;42:1797-803. [ Links ]
9. Bakshi P, Srinivasan R, Rao Kl, et al. Fine needle aspiration biopsy in pediatric space-occupying lesions of liver: a retrospective study evaluating its role and diagnostic efficacy. J Pediatr surg 2006;41:1903-8. [ Links ]
10. Bouyn CI, Leclere J, Raimondo G, et al. Hepatic focal nodular hyperplasia in children previously treated for a solid tumor. Incidence, risk factors, and outcome. cancer 2003;97:3107-13. [ Links ]
11. Yang Y, Fu S, Li A, et al. management and surgical treatment for focal nodular hyperplasia in children. Pediatr surg int 2008;24:699-703. [ Links ]
12. Citake C, Karadeniz C, Oguz A, Boyunaga O, Ekinci O, Okur V. Nodular regenerative hyperplasia and focal nodular hyperplasia of the liver mimicking hepatic metastas is in children with solid tumors and a review of literature. Pediatr hematol oncol 2007;24:281-9. [ Links ]
13. Kumagai H, Masuda T, Oikawa H, Endo K, Endo M, Takano T. Focal nodular hyperplasia of the liver: direct evidence of circulatory disturbances. J gastroenterol hepatol 2000;15:1344-7. [ Links ]
14. Turowski C, Feist h, Alzen G, Gluer s, Petersen C. conversion of a neonatal hepatic hemangioma to focal nodular hyperplasia. Pathol int 2009;59:251-4. [ Links ]
15. Bioulac-sage P, Rebouissou S, Sa Cunha A, et al. Clinical, morphologic, and molecular features defining so-called telangiectatic focal nodular hyperplasias of the liver. Gastroenterology 2005;128:1211-8. [ Links ]
16. Rebouissou S, Bioulac-sage P, Zucman-rossi J. molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J hepatol 2008;48:163-70. [ Links ]
17. Marabelle A, Champagne D, Dechelotte P, Chipponi J, Demeocq F, Kanold J. Focal nodular hyperplasia of the liverin patients previously treated for pediatric neoplastic diseases. J Pediatr hematol oncol 2008;30:546-9. [ Links ]
18. Bioulac-sage P, Balabaud C, Bedossa P, et al. Pathological diagnosis of liver cell adenoma and focal nodular hyperplasia: Bordeaux update. J hepatol 2007;46:521-7. [ Links ]
19. Bioulac-sage P, Cubelg, Balabaud C, Zucman-rossi J. Revisiting the pathology of resected benign hepatocellular nodules using new immune histochemical markers. Semin liver dis 2011;31:91-103. [ Links ]
20. Triantafyllopoulou M, Whitington Pf, Melin-Aldana H, Benya EC, Brickman W. Hepatic adenoma in an adolescent with elevated androgen levels. J Pediatr Gastroenterol nutr 2007;44:640-2. [ Links ]
21. Micchelli ST, Vivekanandan P, Boitnott JK, Pawlik TM, Choti MA, Torbenson M. Malignant transformation of hepatic adenomas. Mod Pathol 2008;21:491-7. [ Links ]
22. Zucman-rossi J, Jeannot E, Nhieu Jt, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with hcc. Hepatology 2006;43:515-24. [ Links ]
23. Grazioli L, Federle MP, Brancatelli G, Ichikawa T, Olivetti L, Blachar A. Hepatic adenomas: imaging and pathologic findings. radiographics 2001;21:877-92. [ Links ]
24. Grazioli L, Federle MP, Ichikawa T, Balzano E, Nalesnik M, Madariaga J. Liver adenomatosis: clinical, histopathologic, and imaging findings in 15 patients. Radiology 2000;216:395-402. [ Links ]
25. Takayasu H, Motoi T, Kanamori Y, et al. two case reports of childhood liver cell adenomas harboring beta-catenin abnormalities. Hum Pathol 2002;33:852-5. [ Links ]
26. Schnater Jm, Kohler Se, Lamers Wh, Von Schweinitz D, Aronson dc. Where do we stand with hepatoblastoma? A review. Cancer 2003;98:668-78. [ Links ]
27. Chung Em, Lattin Ge, Jr., Cube R, et al. From the Archives of the AfiP: pediatric liver masses: radiologic-pathologic correlation part 2. Malignant tumors. Radiographics 2011;31:483-507. [ Links ]
28. De ioris M, Brugieres l, Zimmermann A, et al. Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: the sioPel group experience. Eur J cancer 2008;44:545-50. [ Links ]
29. Herzog CE, Andrassy RJ, Eftekhari F. Childhood cancers: hepatoblastoma. Oncologist 2000;5:445-53. [ Links ]
30. Trobaugh-lotrario Ad, Tomlinson Ge, Finegold MJ, Gore L, Feusner Jh. small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood cancer 2009;52:328-34. [ Links ]
31. Spector Lg, Johnson KJ, Soler Jt, Puumala Se. Perinatal risk factors for hepatoblastoma. Br J cancer 2008;98:1570-3. [ Links ]
32. Oue T, Kubota A, Okuyama H, et al. hepatoblastoma in children of extremely low birth weight: a report from a single perinatal center. J Pediatr surg 2003;38:134-7. [ Links ]
33. Finegold MJ, Egler RA, Goss JA, et al. liver tumors: pediatric population. liver transpl 2008;14:1545-56. [ Links ]
34. Weinberg Ag, Finegold MJ. Primary hepatic tumors of childhood. hum Pathol 1983;14:512-37. [ Links ]
35. Zimmermann A. The emerging family of hepatoblastoma tumours: from ontogenesis to oncogenesis. eur J cancer 2005;41:1503-14. [ Links ]
36. Rowland Jm. hepatoblastoma: assessment of criteria for histologic classification. Med Pediatr oncol 2002;39:478-83. [ Links ]
37. Fuchs J, Rydzynski J, Von Schweinitz D, et al. Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the german cooperative Pediatric liver tumor study HB 94. Cancer 2002;95:172-82. [ Links ]
38. Buendia MA. genetic alterations in hepatoblastoma and hepatocellular carcinoma: common and distinctive aspects. Med Pediatr oncol 2002;39:530-5. [ Links ]
39.Tomlinson GE, Douglass EC, Pollock Bh, Finegold MJ, Schneider NR. Cytogenetic evaluation of a large series of hepatoblastomas: numerical abnormalities with recurring aberrations involving 1q12-q21. Genes chromosomes cancer 2005;44:177-84. [ Links ]
40. Stejskalova E, Malis J, Snajdauf J, et al. cytogenetic and array comparative genomic hybridization analysis of a series of hepatoblastomas. Cancer genet cytogenet 2009;194:82-7. [ Links ]
41. Nagata T, Nakamura M, Shichino H, et al. Cytogenetic abnormalities in hepatoblastoma: report of two new cases and review of the literature suggesting imbalance of chromosomal regions on chromosomes 1, 4, and 12. Cancer genet cytogenet 2005;156:8-13. [ Links ]
42. Surace C, Leszl A, Perilongo G, Rocchi M, Basso G, Sainati l. Fluorescent in situhybridization (fish) reveals frequent and recurrent numerical and structural abnormalitiesin hepatoblastoma with no informative karyotype. Med Pediatr oncol 2002;39:536-9. [ Links ]
43. Von Schweinitz D, Kraus JA, Albrecht S, Koch A, Fuchs J, Pietsch T. Prognostic impact of molecular genetic alterations in hepatoblastoma. Med Pediatr oncol 2002;38:104-8. [ Links ]
44. Yamaoka H, Ohtsu K, Sueda T, Yokoyama T, Hiyama E. diagnostic and prognostic impact of beta-catenin alterations in pediatric liver tumors. oncol rep 2006;15:551-6. [ Links ]
45. Udatsu Y, Kusafuka T, Kuroda S, Miao J, Okada A. high frequency of beta-catenin mutations in hepatoblastoma. Pediatr surg int 2001;17:508-12. [ Links ]
46. Hirschman BA, Pollock BH, Tomlinson GE. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 2005;147:263-6. [ Links ]
47. Pakakasama S, Chen TT, Frawley W, et al. Ccnd1 polymorphism and age of onset of hepatoblastoma. Oncogene 2004;23:4789-92. [ Links ]
48. Eichenmuller M, Gruner I, Hagl B, et al. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009;49:482-90. [ Links ]
49. Adesina Am, Nguyen Y, Guanaratne P, et al. FoXg1 is overexpressed in hepatoblastoma. Hum Pathol 2007;38:400-9. [ Links ]
50. Brotto M, Finegold MJ. Distinct atterns of p27/KIP1gene expression in hepatoblastoma and prognostic implications with correlation before and after Chemotherapy. HumPathol 2002;33:198-205. [ Links ]
51. Shim YH, Park HJ, Choi MS, et al. Hypermethylation of the p16 gene and lack of p16 expression in hepatoblastoma. Mod Pathol 2003;16:430-6. [ Links ]
52. Meyers RL, Rowland Jr, Krailo M, Chen Z, Katzenstein HM, Malogolowkin MH. Predictive power of pretreatment prognostic factors in children with hepatoblastoma: a report from the childrens oncology group. Pediatr Blood cancer 2009;53:1016-22. [ Links ]
53. Roebuck DJ, Perilongo G. Hepatoblastoma: an oncological review. Pediatr radiol 2006;36:183-6. [ Links ]
54. Zimmermann A. Hepatoblastoma with cholangioblastic features (cholangioblastic hepatoblastoma) and other liver tumors with bimodal differentiation in young patients. Med Pediatr oncol 2002;39:487-91. [ Links ]
55. Prokurat A, Kluge P, Kosciesza A, Perek d, Kappeler A, Zimmermann A. transitional liver cell tumors (tlct) in older children and adolescents: A novel group of aggressive hepatic tumors expressing beta-catenin. med Pediatr oncol 2002;39:510-8. [ Links ]
56. Heerema-Mc Kenney A, Leuschner I, Smith N, Sennesh J, Finegold MJ. Nested stromal epithelial tumor of the liver: six cases of a distinctive pediatric neoplasm with frequent calcifications and association with cushing syndrome. Am J surg Pathol 2005;29:10-20. [ Links ]
57. Hill DA, Swanson Pe, Anderson K, et al. Desmoplastic nested spindle cell tumor of liver: report of four cases of a proposed new entity. Am J surg Pathol 2005;29:1-9. [ Links ]
58. Makhlouf HR, Abdul-Al HM, Wang G, Goodman Zd. Calcifying nested stromal-epithelial tumors of the liver: a clinicopathologic, immunohistochemical, and molecular genetic study of 9 cases with a long-term follow-up. Am J surg Pathol 2009;33:976-83. [ Links ]
59. Yu SB, Kim HY, Eo H, et al. Clinical characteristics and prognosis of pediatric hepatocellular carcinoma. World J surg 2006;30:43-50. [ Links ]
60. czauderna P. Adult type vs. Childhood hepatocellular carcinoma -are they the same or different lesions? Biology, natural history, prognosis, and treatment. med Pediatr oncol 2002;39:519-23. [ Links ]
61. Czauderna P, Mackinlay G, Perilongo G, et al. Hepatocellular carcinoma in children: results of the first prospective study of the international society of Pediatric oncology group. J clin oncol 2002;20:2798-804. [ Links ]
62. Wetli SC, Gralla ES, Schibli S, Stranzinger E. Hepatocellular carcinoma and regenerating nodule in a 3-year-old child with Alagille syndrome. Pediatr radiol 2010;40:1696-8. [ Links ]
63. Knisely As, strautnieks Ss, Meier Y, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006;44:478-86. [ Links ]
64. Torbenson M. Review of the clinicopathologic features of fibrolamellar carcinoma. Adv Anat Pathol 2007;14:217-23. [ Links ]
65. El-serag HB, davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A us population-based study. Hepatology 2004;39:798-803. [ Links ]
66. Liu s, chan KW, Wang B, qiao l. fibrolamellar hepatocellular carcinoma. Am J gastroenterol 2009;104:2617-24; quiz 25. [ Links ]
67. Moreno-luna Le, Arrieta O, Garcia-leiva J, et al. clinical and pathologic factors associated with survival in young adult patients with fibrolamellar hepatocarcinoma. Bmc cancer 2005;5:142. [ Links ]
68. Katzenstein HM, Krailo Md, Malogolowkin Mh, et al. fibrolamellar hepatocellular carcinoma in children and adolescents. cancer 2003;97:2006-12. [ Links ]
69. Ward Sc, Waxman S. Fibrolamellar carcinoma: a review with focus on genetics and comparison to other malignant primary liver tumors. semin liver dis 2011;31:61-70. [ Links ]
70. Cruz O, laguna A, Vancells M, Krauel L, Medina M, Mora J. fibrolamellar hepatocellular carcinoma in an infant and literature review. J Pediatr hematol oncol 2008;30:968-71. [ Links ]
71. Stringer MD, Alizai NK. Mesenchymal hamartoma of the liver: a systematic review. J Pediatr surg 2005;40:1681-90. [ Links ]
72. Fukahori S, Tsuru T, Tanikawa K, et al. Mesenchymal hamartoma of the liver accompanied by a daughter nodule: report of a case. surg today 2007;37:811-6. [ Links ]
73. Gow KW, lee l, Pruthi s, Patterson K, healey PJ. mesenchymal hamartoma of the liver. J Pediatr surg 2009;44:468-70. [ Links ]
74. Unal E, Koksal Y, Akcoren Z, Tavl L, Gunel E, Kerimoglu U. mesenchymal hamartoma of the liver mimicking hepatoblastoma. J Pediatr hematol oncol 2008;30:458-60. [ Links ]
75. Hsiao Kh, Lin Lh, Chen Df, Huang Sh. Hepatic mesenchymal hamartoma combined with infantile hepatic hemangioendothelioma in an infant. J formos med Assoc 2007;106:s1-4. [ Links ]
76. Azar Gm, Kutin N, Kahn E. unusual hepatic tumor with features of mesenchymal hamartoma and congenital solitary nonparasitic cyst. Pediatr dev Pathol 2003;6:265-9. [ Links ]
77. Gornicka B, Ziarkiewicz-Wroblewska B, Wroblewski T, et al. Myoid hamartoma of the liver -a novel variant of hamartoma developing in the hilar regionandimitating a malignant liver tumor. med sci monit 2004;10:23-6. [ Links ]
78. Shintaku M, Watanabe K. mesenchymal hamartoma of the liver: a proliferative lesion of possible hepatic stellate cell (ito cell) origin. Pathol res Pract 2010;206:532-6. [ Links ]
79. Osullivan MJ, Swanson Pe, Knoll J, Taboada Em, Dehner lP. undifferentiated embryonal sarcoma with unusual features arising within mesenchymal hamartoma of the liver: report of a case and review of the literature. Pediatr dev Pathol 2001;4:482-9. [ Links ]
80. Begueret H, Trouette H, Vielh P, et al. hepatic undifferentiated embryonal sarcoma: malignant evolution of mesenchymal hamartoma? study of one case with immunohistochemical and flow cytometric emphasis. J hepatol 2001;34:178-9. [ Links ]
81. Narasimhan Kl, Radotra Bd, Harish J, Rao Kl. conservative management of giant hepatic mesenchymal hamartoma. Indian J gastroenterol 2004;23:26. [ Links ]
82. Stocker Jt, Ishak Kg. Undifferentiated (embryonal) sarcoma of the liver: report of 31 cases. cancer 1978;42:336-48. [ Links ]
83. Chowdhary SK, Trehan A, das A, Marwaha RK, Rao Kl. undifferentiated embryonal sarcoma in children: beware of the solitary liver cyst. J Pediatr surg 2004;39:9-12. [ Links ]
84. Iqbal K, Xian Zm, Yuan C. undifferentiated liver sarcoma -rare entity: a case report and review of the literature. J med case reports 2008;2:20. [ Links ]
85. Bisogno G, Pilz T, Perilongo G, et al. undifferentiated sarcoma of the liver in childhood: a curable disease. Cancer 2002;94:252-7. [ Links ]
86. Wei Zg, Tang Lf, Chen Zm, Tang Hf, Li MJ. Childhood undifferentiated embryonal liver sarcoma: clinical features and immunohistochemistry analysis. J Pediatr surg 2008;43:1912-9. [ Links ]
87. Zheng Jm, Tao X, Xu Am, Chen Xf, Wu Mc, Zhang Sh. Primary and recurrent embryonal sarcoma of the liver: clinicopathological and immunohistochemical analysis. Histopathology 2007;51:195-203. [ Links ]
88. Okajima H, Ohya Y, Lee KJ, et al. management of undifferentiated sarcoma of the liver including living donor liver transplantation as a backup procedure. J Pediatr surg 2009;44:33-8. [ Links ]
89. Chuang Wy, Lin Jn, Hung IJ, Hsueh C. undifferentiated sarcoma of the liver. Chang gung med J 2002;25:399-404. [ Links ]
90. Kiani B, Ferrell Ld, Qualman S, Frankel Wl. Immunohistochemical analysis of embryonal sarcoma of the liver. Appl immunohistochem mol morphol 2006;14:193-7. [ Links ]
91. Sowery Rd, Jensen C, Morrison KB, Horsman De, Sorensen Ph, Webber Em. Comparative genomic hybridization detects multiple chromosomal amplifications and deletions in undifferentiated embryonal sarcoma of the liver. cancer genet cytogenet 2001;126:128-33. [ Links ]
92. Rajaram V, Knezevich S, Bove Ke, Perry A, Pfeifer Jd. DNA sequence of the translocation breakpoints in undifferentiated embryonal sarcoma arising in mesenchymal hamartoma of the liver harboring the t(11;19)(q11;q13.4) translocation. genes chromosomes cancer 2007;46:508-13. [ Links ]
93. Kim Dy, Kim Kh, Jung SE, Lee Sc, Park KW, Kim WK. undifferentiated (embryonal) sarcoma of the liver: combination treatment by surgery and chemotherapy.J Pediatrsurg2002;37:1419-23. [ Links ]
94. Uchiyama M, Iwafuchi M, Yagi M, et al. treatment of ruptured undifferentiated sarcoma of the liver in children: a report of two cases and review of the literature. J hepatobiliary Pancreat surg 2001;8:87-91. [ Links ]
95. Das CJ, Dhingra S, Gupta AK, Iyer V, Agarwala S. Imaging of paediatric liver tumours with pathological correlation. Clin radiol 2009;64:1015-25. [ Links ]
96. Wagner Lm, Garrett JK, Ballard Et, et al. Malignant rhabdoid tumor mimicking hepatoblastoma: a case report and literature review. Pediatr dev Pathol 2007;10:409-15. [ Links ]
97. Yuri T, Danbara N, Shikata N, et al. Malignant Rhabdoid tumor of the liver: case report and literature review. Pathol int 2004;54:623-9. [ Links ]
98. Al Nassan A, Sughayer M, Matalka I, et al. INI1 (BAF 47) immunohistochemistry is an essential diagnostic tool for children with hepatic tumors and low alpha fetoprotein. J Pediatr hematol oncol 2010;32:79-81. [ Links ]
99. Marzano E, Lermite E, Nobili C, et al. Malignant rhabdoid tumour of the liver in the young adult: report of first two cases. HPB surg 2009;2009:1-4. [ Links ]
100. Jayaram A, Finegold MJ, Parham Dm, Jasty R. successful management of rhabdoid tumor of the liver. J Pediatr hematol oncol 2007;29:406-8. [ Links ]
101. Machado I, Noguera R, Santonja N, et al. Immunohistochemical study as a tool in differential diagnosis of pediatric malignant rhabdoid tumor. Appl immunohistochem mol morphol 2010;18:150-8. [ Links ]
102. Kuroda H, Moritake H, Sawada K, et al. establishment of a cell line from a malignant rhabdoid tumor of the liver lacking the function of two tumor suppressor genes, Hsnf5/ini1 and p16. Cancer genet cytogenet 2005;158:172-9. [ Links ]
103. Christison-Lagay Er, Burrows Pe, Alomari A, et al. hepatic hemangiomas: subtype classification and development of a clinical practice algorithm and registry. J Pediatr surg 2007;42:62-7. [ Links ]
104. Emre S, McKenna GJ. Liver tumors in children. Pediatr transplant 2004;8:632-8. [ Links ]
105. Sari N, Yalcin B, Akyuz C, Haliloglu M, Buyukpamukcu M. infantile hepatic hemangioendothelioma with elevated serum alpha-fetoprotein. Pediatr hematol oncol 2006;23:639-47. [ Links ]
106. Dong Kr, Zheng S, Xiao X. conservative management of neonatal hepatic hemangioma: a report from one institute. Pediatr surg int 2009;25:493-8. [ Links ]
107. Mo Jq, Dimashkieh Hh, Bove Ke. GLUT1 endothelial reactivity distinguishes hepatic infantile hemangioma from congenital hepatic vascular malformation with associated capillary proliferation. Hum Pathol 2004;35:200-9. [ Links ]
108. Huang SA, Tu HM, Harney JW, et al. severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J med 2000;343:185-9. [ Links ]
109. Konrad D, Ellis G, Perlman K. Spontaneous regression of severe acquired infantile hypothyroidism associated with multiple liver hemangiomas. Pediatrics 2003;112:1424-6. [ Links ]
110. Bessho K, Etani Y, Ichimori H, et al. Increased type 3 iodothyronine deiodinase activity in a regrown hepatic hemangioma with consumptive hypothyroidism. Eur J Pediatr 2010;169:215-21. [ Links ]
111. Sharif K, English M, Ramani P, et al. management of hepatic epithelioid haemangio-endothelioma in children: what option? Br J cancer 2004;90:1498-501. [ Links ]
112. Da ines D, Petitcolin V, Joubert-Zakeyh J, Demeocq F, Garcier Jm. Epithelioid hemangioendothelioma of the liver with metastatic coeliac lymph nodes in an 11-year-old boy. Pediatr radiol 2010;40:1293-6. [ Links ]
113. Dimashkieh Hh, Mo Jq, Wyatt-Ashmead J, Collins Mh. Pediatric hepatic angiosarcoma: case report and review of the literature. Pediatr dev Pathol 2004;7:527-32. [ Links ]
114. Burrows Pe, Dubois J, Kassarjian A. Pediatric hepatic vascular anomalies. Pediatr radiol 2001;31:533-45. [ Links ]
115. Nord Km, Kande lJ, Lefkowitch Jh, et al. Multiplecutaneous infantile hemangiomas associated with hepatic angiosarcoma: case reportand review ofthe literature. Pediatrics2006;118:907-13. [ Links ]
116. Nazir Z, Pervez S. Malignant vascular tumors of liver in neonates. J Pediatr surg 2006;41:49-51. [ Links ]
117. Ganguly R, Mukherjee A. Infantile hemangioendothelioma: A case report and discussion. Pathol res Pract 2010;206:53-8. [ Links ]
Ana Rodrigues
Serviço de Anatomia Patológica, Faculdade de Medicina da Universidade do Porto
Alameda Professor Hernâni Monteiro, 4200 Porto
Email: med06170@med.up.pt

