










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.27 no.5 Porto out. 2013
ARTIGO DE REVISÃO
Colangite esclerosante primária e colite ulcerosa: factos e incertezas
Primary sclerosing cholangitis and ulcerative colitis: Facts and uncertainties
Gonçalo Esteves1, Fátima Carneiro2
1 Faculdade de Medicina da Universidade do Porto, Porto
2 Serviço de Anatomia Patológica, Centro Hospitalar de São João e Faculdade de Medicina da Universidade do Porto, e IPATIMUP, Porto
RESUMO
Há uma relação descrita, embora ainda mal esclarecida, entre a colangite esclerosante primária e a doença inflamatória intestinal, partiCUlarmente a colite ulcerosa. A concomitância de ambas as patologias encontra na última algumas alterações fenotípicas à sua apresentação isolada, dita clássica. Acompanhando essas alterações, constatam-se também diferentes impactos na vida destes doentes, por exemplo, em relação ao risco de neoplasia. Há controvérsia quanto à legitimidade de se considerar dois subgrupos de colangite esclerosante primária: um associado à colite ulcerosa e outro isolado. Têm-se procurado, ainda sem respostas cabais, formas de diagnóstico desta associação de doenças através de autoanticorpos e de genes de suscetibilidade. É intenção deste trabalho reunir o conhecimento atual nesta área da Gastroenterologia e da Hepatologia.
Palavras-chave: Colangite esclerosante, doença inflamatória intestinal, colite ulcerosa, carcinoma coloretal
ABSTRACT
Primary sclerosing cholangitis and inflammatory bowel disease, mainly ulcerative colitis, are in some way interconnected. The latter presents some phenotypical changes to its classical presentation when associated with the former. Besides, there are additional demands when handling patients with these diseases, as reflected for instance by the distinct risk of neoplasia, which is difficult to ascertain. The splitting of primary sclerosing cholangitis in two groups, a colitis- associated one and an isolated one, is still controversial. lately there has been investigation on how to diagnose these associated diseases through autoantibodies titres and susceptibility genes, but no straightforward results were reached yet. Here we assemble the up-to-date data on this Gastroenterology/Hepatology issue.
Key-words: Sclerosing cholangitis, inflammatory bowel diseases, ulcerative colitis, colorectal cancer
INTRODUÇÃO
A colangite esclerosante primária(CEP) é uma doença inflamatória crónica rara que atinge os canais biliares intra e extra-hepáticos de médio e grande calibre, com sua progressiva fibrose e estenose. Na maior parte dos indivíduos afetados, culmina em cirrose, hipertensão portal e descompensação hepática, uma vez que ainda não estão disponíveis terapêuticas de eficácia comprovada, além do transplante hepático.1 A assinalar ainda, o contexto neoplásico é delicado: além do risco óbvio de colangiocarcinoma, avaliado em 160 vezes o da população geral, estes doentes estão sujeitos a um risco acrescido de carcinoma hepatocelular, da vesícula biliare dopâncreas.2
A etiologia precisa da CEP é desconhecida (diagnostica-se geralmente por exclusão), mas é provável que lhe subjaza um mecanismo imunopatogénico, dadas as associações encontradas com doenças autoimunes, com a doença inflamatória intestinal (DII) e com haplótipos do HLA (Human Leukocyte Antigen).3 Não será, todavia, uma doença autoimune clássica, visto afetar duas vezes mais homens do que mulheres e não responder à terapêutica imunossupressora como tal.4 Há autores a defenderem o contrário, porquanto a razão homens/mulheres é inferior (i.e. compatível com a noção de doença autoimune clássica) no grupo de doentes com CEP não associada à colite ulcerosa, que, embora atingindo uma minoria, é por aqueles considerada a verdadeira CEP.5
A colite ulcerosa (CU) é a mais prevalente das DII, encontrando-se as incidências e prevalências mais elevadas na América do norte e no norte da europa.6 Além dos sintomas gastrointestinais próprios, o diagnóstico é confirmado pelos exames endoscópico e histológico: trata-se de uma situação inflamatória caracteristicamente restrita à mucosa, com início retal e progressão proximal contínua. Em função da distribuição anatómica da doença, podemos dividi-la em três grupos: proctite, colite esquerda e pancolite, com ou sem ileíte retrógrada (vide infra A díada CEP/CU).6 Veja-se que, enquanto o reconhecido risco de carcinoma colorretal na CU se relaciona diretamente com a extensão e o tempo de atividade da doença, na CEP, a relação temporal com o risco de colangiocarcinoma não se verifica, pois até 50% dos doentes diagnosticados com esta neoplasia são-no durante o primeiro ano após o diagnóstico de CEP.7
Na fisiopatologia da CU, o aumento da permeabilidade da barreira intestinal, seja ele causa ou consequência da doença, permite um contacto mais desimpedido entre as células da lâmina própria e os antigénios luminais. A ativação da imunidade inata (i.e. macrófagos e células dendríticas) em primeira instância e da imunidade adaptativa numa segunda fase é uma explicação provável para o início e perpetuação da inflamação intestinal.6
Pensa-se que a existência de uma associação entre CEP e CU possa apontar para uma patogénese comum, e é nessa expectativa que tem avançado o seu estudo. Nas seguintes secções, reveem-se os principais estudos no âmbito desta associação patológica.
MÉTODO
Este trabalho resultou de uma pesquisa na base de dados PubMed/MEDLINE (http://www.ncbi.nlm.nih.gov/pubmed/) entre dezembro de 2011 e dezembro de 2012. Os descritores usados foram primary sclerosing cholangitis, inflammatory bowel disease e ulcerative colitis, de que resultou um total de 681 artigos. Foram incluídos os trabalhos escritos em língua inglesa que tratassem de uma e/ ou de outra patologia (CEP e DII) sem descurar o quadro de sobreposição clínica das duas e desenvolvendo as seguintes áreas: (a) epidemiologia, (b) anatomia patológica, (c) fisiopatologia, (d) autoimunidade e (e) genética. Excluímos desta revisão os estudos pediátricos, bem como os centrados na terapêutica destas patologias.
A DÍADA CEP/CU
A CEP é a doença hepatobiliar que mais vezes se associa à DII.8 Em populações ocidentais (europeias e americanas), cerca de70 a 80% dos doentes com CEP padece de uma DII, que é a CU em até 86% dos casos.9 note-se que há exceções e variações geográficas consideráveis nesta tendência:10 por exemplo, no Japão, o valor pode ser tão baixo quanto 21%.11 o contrário, porém, não é verdade: estima-se que apenas cerca de 5% dos doentes com CU tenham CEP em simultâneo.10 esta desigualdade é facilmente aceite se considerarmos que o número de doentes com CU supera largamente o número de casos de CEP (Fig.1).
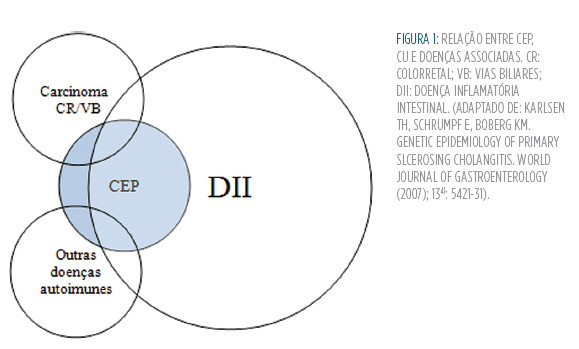
Atualmente, não é possível estabelecer a CEP associada à CU como uma entidade nosológica diferente da CEP isolada.12 em 1990, Rabinovitz et al estudaram um grupo de 47 doentes com CEP e CU (19 controlos com CEP isolada): o género masculino era predominante, a apresentação mais frequente era por alteração das provas de função hepática (sendo a fadiga, o prurido e a icterícia mais comuns na CEP isolada) e observavam-se mais vezes estenoses combinadas intra e extra-hepáticas (ao contrário dos controlos, em que era mais frequente o envolvimento extra-hepático único).13 Contudo, não foram encontradas diferenças por outros estudos de que tenhamos conhecimento.
Os sintomas inflamatórios intestinais dos doentes com CEP e CU antecedem, em geral, vários anos as manifestações e o diagnóstico da CEP, embora o contrário também possa suceder, comum intervalo de até 4 anos.5 Sendo as linfadenopatias peri-hepáticas achados ecográficos característicos da CEP, a sua presença pode ser um indicador altamente preditivo de CEP num doente com CU (mais do que os parâmetros séricos isoladamente).14 É de assinalar que a CEP diagnosticada em indivíduos com CU prévia se encontra, em regra, numa fase muito inicial da sua evolução, dada por uma pontuação de risco Mayo (fatores: idade, níveis de bilirrubina, albumina e aminotransférase do aspartato e história de rotura de varizes esofágicas15) mais baixa do que na CEP isolada à data do diagnóstico. Duas hipóteses surgem: ou estamos perante um viéstemporal (leadtimebias) – os doentes com CU são avaliados com frequência, ao contrário dos indivíduos saudáveis até então – ou há um substrato biológico intrínseco que justifica as diferentes apresentações da doença consoante o contexto patológico do doente.16 Voltaremos à questão genética mais adiante.
A CU associada à CEP (Tabela 1) tem sido descrita classicamente, e sempre em contraponto à sua forma isolada, como uma DII de menor atividade inflamatória, com alta prevalência de pancolite, atingimento preferencial do cólon direito, sem atividade retal e com apresentação de um fenómeno incomum, a ileíte retrógrada (backwash ileitis) [9]. Estudos recentes subvertem, contudo, parte destas assunções. Em 2009, Joo et al (coreia), tendo verificado uma prevalência de pancolite superior no grupo CEP/CU (85% vs. 45%; p <0,05), não encontraram diferenças quanto à ileíte retrógrada e ao atingimento retal.17 Na mesma linha se encontra o estudo de Boonstra et al (Holanda, 2012), que, dispondo de uma das maiores coortes populacionais de doentes com CEP de que tivemos conhecimento até hoje, demonstraram, no grupo CEP/CU, uma prevalência de pancolite de 94% e um predomínio da inflamação do cólon direito, classificando a ileíte retrógrada e o não atingimento retal como achados raros.18 Note-se que estes estudos definem ileíte retrógrada com ligeiras diferenças entre si.

Um estudo japonês recente salienta que os doentes com CEP e CU são significativamente mais jovens do que os doentes com CEP isolada (média das ida des: 33,6 vs. 58,9; p <0,001) e que, entre os doentes com CU, os que sofrem menos exacerbações reque rendo internamento são os que simultaneamente padecem de CEP.19 Estes resultados reiteram o caráter mais ligeiro da DII destes indivíduos, quando comparados com os doentes sem CEP, medido pela menor necessidade de imunossupressores (incidências cumulativas a 10 anos: 24% vs. 46%; p <0,001) e de intervenção cirúrgica (indicação para resseção: 17,3% vs. 36,7%; p <0,003) e pelo maior consumo de ácido 5-aminossalicílico (5-ASA).20 Porém, constatou-se que, nos doentes com CEP e CU, o risco de displasia e carcinoma colorretal (CCR) é superior ao dos doentes com CU isolada.21 Sendo a extensão e a duração da doença intestinal os principais fatores determinantes do seu comportamento e prognóstico, é discutível a legitimidade de se estabelecer essa comparação, já que nos primeiros a prevalência de pancolite é mais elevada. Não obstante, há que considerar os seguintes dados: uma metanálise demonstrou um risco de CCR quatro vezes superior no grupo CEP/CU, em relação ao de CU isolada,22 e um estudo do mesmo ano encontrou um risco no primeiro grupo dez vezes superior ao da população geral.2 Broomé et al (1995) estimaram, nos doentes CEP/CU, as incidências cumulativas de displasia/CCR a 10, 20 e 25 anos em 9%, 31% e 50%, em contraposição a 2%, 5% e 10%, respetivamente, nos doentes com CU isolada (p <0,001).23 Estes números apelam à prudência do médico, que deve reconhecer o substancial risco de displasia e CCR a que aqueles doentes podem estar sujeitos e submetê-los a um programa de controlo colonoscópico assim que a CEP seja diagnosticada.12 As linhas orientadoras da American Association for the Study of Liver Diseases recomendam que o rastreio se inicie logo que diagnosticada a CEP, independentemente da duração da CU, com uma periodicidade de 1 a 2 anos.1 É possível que o desenvolvimento de CEP induza, em doentes com CU, um estado inicial de indolência clínica e que neste esteja implicado algum fator genético ou imunológico diretamente relacionado com a CEP, podendo ser a causa do maior risco neoplásico nestes doentes.24 Apesar das hipóteses, ainda se desconhece a justificação deste risco acrescido, que merece mais investigação.
Quanto ao prognóstico de longo termo dos doentes com CEP e CU e com CEP isolada (i.e. necessidade de transplante hepático, risco de cancerização e mortalidade), os resultados dos estudos mais recentes são contraditórios.16,25 um estudo neozelandês – dispondo de um número limitado de doentes com CEP (sem DII) no grupo controlo – mostrou que os doentes do grupo CEP/CU são mais frequentemente alvo de transplante hepático (p <0,03) e de complicações malignas (p<0,017).25 Em contrapartida, U. Navaneethan et al (E.U.A.) concluíram, ajustando a pontuação de risco Mayo, a duração da CEP e o género dos doentes nos dois grupos, que a CU não influi na necessidade de transplante nem tem impacto na mortalidade associada à CEP.16
É interessante verificar que há uma aparente dissociação da gravidade de apresentação clínica destas duas patologias. Doentes com CEP grave e progressiva que requeira transplante têm cursos mais ligeiros da DII e, por outro lado, doentes com CU que exija colectomia será improvável virem a precisar de transplante hepático para o tratamento da CEP.26,27 Outra observação que reforça esta hipótese é a de que a CEP pode surgir vários anos após colectomia total por DII de difícil controlo.5 No que diz respeito aos efeitos da colectomia sobre a incidência de CEP recidivada, alguns estudos demonstraram que a colectomia realizada antes ou durante (não após) o transplante hepático tem um efeito protetor da recidivapós-transplante da CEP.28-30 A sobrevida a 5 anos desses doentes, todavia, não difere da dos transplantados sem colectomia prévia (86%emambos os grupos).31 Em 2012, um estudo norueguês verificou que o transplante hepático resulta numa melhoria da atividade da DII (42 casos)32 – o que temos que admitir poder dever-se à imunossupressão –, enquanto uma análise retrospetiva húngara do mesmo ano (31 casos) observou o oposto, apoiando a realização de colectomia a priori.33 Acrescente-se ainda que os doentes com CEP, após colectomia com bolsa ileoanal por CU, parecem sofrer, mais vezes do que os doentes sem doença hepática, inflamação da anastomose (pouchitis)18,34,35 e ileíte préanastomótica.36
Em suma, por um lado, a CEP recidiva mais vezes no transplante quando há inflamação intestinal ativa,37 e, por outro, a atividade da DII pode ser atenuada pelo transplante hepático.
Em relação ao risco de CCR em doentes transplantados, vera et al (reino unido, 2003) fixaram as incidências cumulativas a 5 e 10 anos em 14% e 17%, respetivamente, contrastando com 0% a 10 anos no grupo com CEP isolada (p <0,06).38 O risco de CCR pode estar inicialmente aumentado em virtude da agressiva imunossupressão prescrita após o transplante, como foi reforçado por um grupo holandês no mesmo ano.39 Contrariando estes e outros estudos prévios, Dvorchik et al demonstraram que o transplante hepático acelera a progressão da DII, muito embora não aumente o risco de CCR além do nível determinado pela duração da DII.40 A presença de CU parece não afetar o risco de colangiocarcinoma na CEP.41
PATOGÉESE DA CEP E DA CEP/CU
Algumas hipóteses têm sido avançadas para explicar a patogénese da CEP. Dada a associação com a DII, Vierling (1998) propôs uma explicação unificadora: antigénios bacterianos provenientes do cólon atingiriam a árvore biliar através da circulação portal, devido à permeabilidade aumentada do epitélio intestinal, e aí mimetizariam moléculas dos colangiócitos.42 As células de Kupffer seriam induzidas a produzir citocinas, atraindo neutrófilos, macrófagos, linfócitos e fibroblastos, e conduzindo a longo prazo às lesões de atrofia, colestase, isquemia, fibrose e cirrose que caracterizam a doença.43 Não se explica assim, contudo, a razão porque há menos doentes com doença de Crohn (DC) do que com CU associada à CEP e, ao mesmo tempo, por que é que a CEP e a DII seguem muitas vezes cursos independentes, como se viu acima. Não está comprovado que o sobrecrescimento bacteriano em humanos leve à CEP,44 embora em ratinhos geneticamente suscetíveis isso pareça ser uma realidade.45 Na CU, ao contrário, estudos em humanos têm atribuído à flora intestinal um papel fulcral não apenas na patogénese, como também no nível de inflamação e na definição do fenótipo da doença (CU ou DC).6
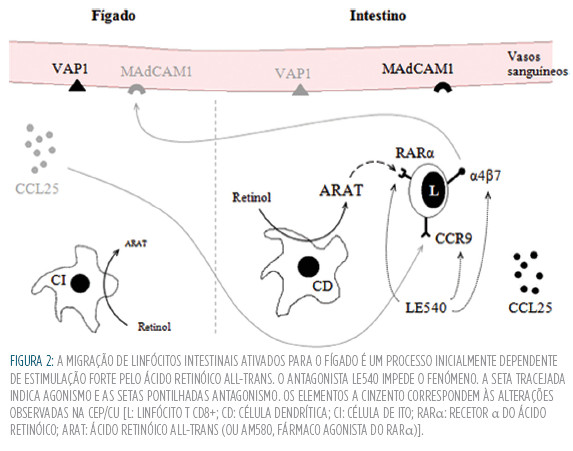
Outros autores tentam explicar a distribuição de linfócitos entre o intestino e o fígado. Em 2002, Grant et al sugeriram que células TDE memória originalmente ativadas no intestino podem atingir o fígado e aí perpetrar uma reação inflamatória, devido à expressão aberrante de adesinas e quimiocinas que em condições normais seriam exclusivas do intestino46 (Fig.2). A adressina MAdCAM1 (Mucosal Addressin Cell Adhesion Molecule 1), expressa em regra apenas nos vasos da mucosa intestinal, pode surgir no endotélio hepático de doentes com patologia inflamatória crónica do fígado,47 assim como a quimiocina CCL25.48 Os linfócitos intestinais exprimem na sua superfície celular as moléculas a4ß7 (ligando da MAdCAM1) e CCR9 (recetor da CCL25), que são produzidas em resposta às células dendríticas locais, por meio de um processo dependente do ácido retinóico (AR).49 Apesar de o fígado dispor de uma reserva de AR nas células de ito, as suas células dendríticas não são capazes de gerar um sinal suficientemente forte (transformar bastante retinol em ácido retinóico all-trans) para induzir a síntese de a4ß7 e CCR9 nos linfócitos, de forma a explicar a presença destes fenótipos moleculares no fígado.49 Daqui se conclui que estas células são ativadas no intestino, e só então recrutadas para o fígado, através da circulação entero-hepática, em resposta à expressão ectópica da MAdCAM1 e da CCL25.50 Por sua vez, a VAP1 (Vascular Adhesion Protein 1), que medeia a adesão linfocitária e está constitutivamente expressa no endotélio hepático, encontra-se sobre-expressa no endotélio intestinal na DII.51 Mais uma vez fica por justificar a preferência de associação da CEP com a CU, em detrimento da dc. Já os cursos independentes que aquelas duas doenças muitas vezes tomam podem ser por este meio explicados, visto que um mesmo grupo de linfócitos eventualmente comum às duas patologias poderia circular entre um e outro órgão, fomentando aí a atividade inflamatória em função dos níveis de citocinas presentes.27
Ainda há outra explicação que pretende assemelhar a CEP à aterosclerose: a incapacidade relativa de os colangiócitos transportarem fosfolipídeos para o lúmen biliar submetê-los-ia à toxicidade oxidativa direta dos ácidos biliares e da bile sobressaturada em colesterol, gerando lesões de esclerose, o que foi verificado apenas em modelos animais knockout para o Mdr2 (Multidrug resistance 2).52 Esta hipótese, todavia, não considera a DII que acompanha, na maioria dos casos, esta doença. Os modelos Mrd2 -/-não desenvolvem DII.52
IMUNOPATOLOGIA
Um dos argumentos a favor de se considerar a CEP uma doença autoimune é a abundância de autoanticorpos em circulação. Anticorpos citoplasmáticos antineutrofílicos atípicos de padrão perinuclear (p-ANCA daqui em diante) circulam em 33-88% dos doentes com CEP, em 60-87% dos doentes com CU e em 50-96% dos doentes com hepatites autoimunes.4 Não sendo ferramentas diagnósticas de nenhuma destas patologias, é de notar que estão mais intimamente associados à CU do que à dc (44,1% vs. 8,1%; p <0,0002),53 podendo naquela funcionar como um teste altamente específico (98%), embora pouco sensível (42%).54 O título de p-ANCA parece correlacionar-se, na CU, com a atividade da doença (p <0,05) e, nestes doentes, os p-ANCA têm mais frequentemente dois ou mais antigénios-alvo do que na DC (p <0,01), segundo um estudo recente.55 A positividade54 e a alta concentração55 de p-ANCA associam-se ainda a CU de longa duração (p <0,05), e curiosamente também à CU de predomínio no cólon esquerdo (p <0,001).55
A BPI (bactericidal/permeability-increasing protein) foi proposta como alvo possível dos p-ANCA [56]. Trata-se da proteína dos grânulos neutrofílicos com atividade antimicrobiana mais consistente, em particular contra bactérias negativas em coloração de Gram, nas quais tem um efeito potencialmente neutralizador da toxicidade dos lipopolissacarídeos da sua parede.57,58 Stoffel et al (1996) investigaram essa hipótese e verificaram que a BPI surgia como antigénio-alvo principal dos p-ANCA em 44% dos doentes com CEP e em 38% dos doentes com CU.56 Este resultado foi corroborado na CU por um grupo australiano (2000)59 e na CEP por um grupo holandês (1998) que também observou uma associação entre os p-ANCA anti-BPI e anticatepsina G e a presença de cirrose hepática, sugerindo uma evolução mais agressiva da doença nesse contexto.60 Note-se, contudo, que os autoanticorpos anti-BPI são também detetáveis noutras patologias, como em vasculites e na fibrose cística.61

Ainda que a lactoferrina – outra proteína dos grânulos neutrofílicos com ação antibacteriana62 – não aparente ser um alvo antigénico preferencial dos p-ANCA na CEP e em outras doenças hepáticas autoimunes (hepatite autoimune, cirrosebiliar primária, colangite autoimune), proporções semelhantes destes doentes (rondando os 30%) partilham uma perda de tolerância à mesma.63 A prevalência dos anticorpos antilactoferrina na CEP (4-54%) é próxima da verificada na CU (4-50%), e consideravelmente superior à da dc (0-9%).64 Roozendaal et al mostraram que estes p-ANCA são mais frequentemente detetados na díada CEP/CU do que na CEP isolada.60
Numa série de 15 indivíduos com CEP, Orth et al (1998) detetaram anticorpos anticatálase (ACC) em 9 doentes (60%).65 Estes anticorpos, dirigindo-se a uma enzima antioxidante dos peroxissomas, fazem supor que o stress oxidativo possa contribuir para a patogénese da CEP. No mesmo estudo observou-se que os doentes com CEP e ACC têm um curso mais grave da doença, com um doseamento de fosfátase alcalina significativamente superior, do que os doentes com CEP sem ACC detetável. Ainda mais, uma vez que, nos doentes com apenas CU, os ACC não foram doseáveis, concluiu-se que a DII não é responsável pelo surgimento de ACC em doentes com CEP e CU em simultâneo.65
Terjung et al (2000) demonstraram, em 92% dos doentes analisados (com CU, CEP ou hepatite autoimune), que os p-ANCA se ligavam a uma proteína mielóide de 50 kilodalton do invólucro nuclear.66 Em 2005 e 2010, dois grupos alemães encabeçados pelo mesmo investigador apontaram, respetivamente, o isotipo 5 da tubulina ß (TB5) como sendo o autoantigénio em causa67 e a proteína bacteriana ftsZ com o alvo de reação cruzada dos p-ANCA nas doenças hepáticas autoimunes (TB5: 72-88%; ftsZ: 64-82%).68 Este elemento do citosqueleto, precursor evolutivo da tubulina e presente em quase todas as bactérias da flora intestinal, foi assim descrito pela primeira vez na Escherichia coli, e forma uma estrutura anelar submembranar no plano equatorial da célula que participa no fenómeno de cissiparidade.69,70 demonstrou-se também, em modelos animais de DII (ratinhosil10 -/-), que os anticorpos anti-TB5 são sintetizados em resposta a microrganismos intestinais.68 Estas evidências apoiam a hipótese de um cólon hiperpermeável na patogénese da CEP associada à CU (vide supra a hipótese de vierling).71
SUSCETIBILIDADE GENÉTICA
Alguns dados epidemiológicos fazem crer que a CEP tenha um forte componente genético na sua base: irmãos de indivíduos com CEP desenvolvem esta doença com uma frequência 9 a 39 vezes superior à da população geral e têm um risco de desenvolver CU 8 vezes aumentado.72 Esta associação estreita a relação da díada CEP/CU.
A CEP é uma doença geneticamente complexa, isto é, não segue os padrões de transmissão mendelianos, tendo já sido identificados vários loci do sistema HLA (Human Leukocyte Antigen) que conferem suscetibilidade à doença. A CU não fica de todo atrás, com mais de 30loci de risco encontrados só na última década.
As primeiras associações genéticas da CEP, propostas por um grupo norueguês, datam de 1982 e envolvem os subtipos dr3 e B8 do HLA.73 Trata-se de polimorfismos de um nucleotídeo (single nucleotide polymorphisms, SNP) no cromossoma 6p21 que ultimamente foram alvo de duas amplas análises de associação genómica, verificando-se uma ligação mais forte da CEP com a região do HLA-B.74,75 Na CU, porém, a associação ao HLA é mais modesta, sendo os SNP encontrados mais em relação com os genes da classe II do mesmo.75 Não é despiciendo sublinhar que, na região do complexo HLA, genes vizinhos estão relacionados de forma íntima (mais do que a aleatoriedade da segregação cromossómica faria supor) – num considerável desequilíbrio de ligação (linkage disequilibrium) – e, por conseguinte, torna-se muito difícil definir quais os exatos genes de suscetibilidade em causa neste setor do genoma.76
No que se refere ao restante genoma (para além do complexo HLA), foram já identificados os seguintes loci: 2q13, 2p16, 2q35, 3p21, 4q27, 9q34, 10p15 e 13q31.74,75,77 Destes, os cinco que também se associam à CU são abordados a seguir (Tabela 1).
No locus 2p16, um possível candidato a gene de suscetibilidade é o REL, que codifica um mediador importante da via de sinalização do NF-kB (Nuclear Factor kB).77 No 2q35, o gene proposto é o GPBAR1 (mais conhecido por recetor de ácidos biliares TGR5).74 Este é um candidato muito interessante, uma vez que o recetor codificado parece concorrer para a regulação da integridade da barreira intestinal. Ficou demonstrado que, em ratinhos knockout GPBAR1 -/-,existe uma perturbação da arquitetura molecular das junções apertadas do epitélio cólico, o que culmina num aumento da permeabilidade intestinal, tornando os animais especialmente sensíveis ao desenvolvimento de colite quando expostos a um agente agressor da mucosa.78 No cromossoma 3p21, é provável que o gene MST1 (Macrophage-Stimulating 1), cujo produto tem elevados níveis de expressão no epitélio da vesícula biliar e está envolvido na imunidade inata, seja um locus de suscetibilidade quer para a CEP, quer para a DII, sendo de realçar a substituição R689C, que compromete a interação da proteína com o seu recetor.79 Dado o forte desequilíbrio de ligação também presente entre os genes desta região, é difícil descartar um eventual papel de outros genes, como o da peroxídase 1 do glutatião.75,79 Finalmente, quanto aos loci 4q27 e 9q34,de entre os vários genes que podem ser nomeados, sobressaem, respetivamente, os das interleucinas 2 e 21 (IL2 e IL21) e o CARD9.77 A constatação de que ratinhos knockout IL2RA (recetor a da IL2) -/-desenvolvem espontaneamente inflamação intestinal e biliar leva a crer que a via da IL2 esteja implicada na patogénese da CEP e da CU.80 A proteína resultante do CARD9 estimula a imunidade inata em resposta a agentes patogénicos intra e extra celulares, mais concretamente promovendo a diferenciação de células T helper do tipo 17 e a produção de citocinas pró-inflamatórias.81
CONCLUSÕES
As evidências epidemiológicas, clínicas, anátomopatológicas, imunológicas e genéticas que temos vindo a descrever sugerem-nos alguns pontos de confluência da CEP e da CU, mas muitas delas não são completamente esclarecedoras e parecem contradizer-se, como pudemos apreciar na secção A díada CEP/CU. Veja-se que os trabalhos que sustentam estas considerações são em regra retrospetivos, com as limitações que se lhes atribuem.
Aquela que durante anos tinha sido a apresentação clínica típica da CU com as particularidades próprias de um doente com CEP foi ultimamente desacreditada por estudos que apenas reconheceram a maior extensão da doença intestinal e a sua menor atividade inflamatória neste contexto.
A seleção de uma coorte de doentes com CEP e CU de fenótipo clássico (i.e. sem diferenças significativas em relação à CU isolada) pode auxiliar na confirmação e estabelecimento do risco neoplásico da CU associada à CEP – mitigando, deste modo, o enviesamento conferido por uma DII mais extensa e de maior duração. Este é um aspeto clínico que seria útil aferir, já que a sua gravidade tem um impacto negativo na qualidade de vida dos doentes.
A respeito da intensidade inflamatória das duas patologias, a sua relação de aparente desigualdade, podendo advir das movimentações de um mesmo conjunto de linfócitos (vide supra em Patogénese da CEP e da CEP/CU), permite-nos aceitar que, quer após transplante hepático, quer após colectomia, a doença recidive no novo órgão ou na bolsa ileoanal, respetivamente, tornando-se a outra mais fruste. É possível, portanto, conciliar os mecanismos patogénicos propostos com as observações clínicas.
No terreno da autoimunidade, a situação é mais nebulosa, com os estudos a apontarem em direções diferentes, com resultados díspares. Nenhum dos p-ANCA até hoje descritos está associado a qualquer das duas doenças ou à díada em exclusividade, embora os seus alvos possam, por outro lado, orientar-nos na clarificação da sua patogénese. Muitos dos antigénios em destaque são proteínas bacterianas ou proteínas envolvidas na resposta antibacteriana do hospedeiro. Poderá ser conveniente avaliar a aplicabilidade de um determinado conjunto de autoanticorpos com força de associação suficiente para servir de rastreio em indivíduos com CU em risco de desenvolverem CEP, ou vice-versa.
O facto de apenas dois loci de suscetibilidade, de entre os quinze hoje estabelecidos na CU, terem sido associados significativamente à CEP (a saber: 2q35 e 3p21)74 pode apoiar a teoria de que a inflamação intestinal na CEP representa um fenótipo distinto de DII. Mais estudos genéticos neste âmbito estão em curso. Principalmente para a CEP, em que nenhuma terapêutica médica disponível interfere na progressão para cirrose e transplante,1 parece-nos relevante a exploração genética – por exemplo a fim de se perceber o real papel dos ácidos biliares na inflamação biliar (e intestinal) – pois pode vir a ter implicações terapêuticas. Seria também interessante procurar explicar geneticamente as variações geográficas da associação CEP/CU.
REFERÊNCIAS
1. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, Gores GJ. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51(2):660-78. [ Links ]
2. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Loof L, Danielsson A, Hultcrantz R, Lindgren S, Prytz H, Sandberg-Gertzen H, Almer S, Granath F, Broomé U. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol 2002;36(3):321-7. [ Links ]
3. Pollheimer MJ, Halilbasic E, Fickert P, Trauner M. Pathogenesis of primary sclerosing cholangitis. Best Pract res Clin Gastroenterol 2011;25(6):727-39. [ Links ]
4. Chapman R, Cullen S. Etiopathogenesis of primary sclerosing cholangitis. World J Gastroenterol 2008;14(21):3350-9. [ Links ]
5. Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis 1991;11(1):31-9. [ Links ]
6. Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet 2012;380(9853):1606-19. [ Links ]
7. Razumilava N, Gores GJ, Lindor Kd. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology 2011;54(5):1842-52. [ Links ]
8. Bernstein CN, Blanchard JF, Rawsthorne P, YU N. The prevalence of extraintestinal diseases in inflammatory bowel disease: a population-based study. Am J Gastroenterol 2001;96(4):1116-22. [ Links ]
9. Loftus Jr. EV , Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR, Jewell DA, Sandborn WJ. PSC - IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005;54(1):91-6. [ Links ]
10. Saich R, Chapman R. Primary sclerosing cholangitis, autoimmune hepatitis and overlap syndromes in inflammatory bowel disease. World J Gastroenterol 2008;14(3):331-7. [ Links ]
11. Takikawa H,Takamori Y, Tanaka A, Kurihara H, Nakanuma Y. Analysis of 388 cases of primary sclerosing cholangitis in Japan; presence of a subgroup without pancreatic involvement inolder patients. Hepatol res 2004;29(3):153-9. [ Links ]
12. Broomé U, Bergquist A. Primary sclerosing cholangitis, inflammatory bowel disease, and colon cancer. Semin Liver Dis 2006;26(1):31-41. [ Links ]
13. Rabinovitz M, Gavaler JS, Schade RR , Dindzans VJ, Chien MC, Van Thiel DH. Does primary sclerosing cholangitis occurring in association with inflammatory bowel disease differ from that occurring in the absence of inflammatory bowel disease? A study of sixty-six subjects. Hepatology 1990;11(1):7-11. [ Links ]
14. Hirche To, Russler J, Braden B, Schuessler G, Zeuzem S, Wehrmann T, Seifert H, Dietrich CF. Sonographic detection of perihepatic lymphadenopathy is an indicator for primary sclerosing cholangitis in patients with inflammatory bowel disease. Int J Colorectal Dis 2004;19(6):586-94. [ Links ]
15. Kim WR, Therneau TM, Wiesner RH, Poterucha JJ, Benson JT, Malinchoc M, Larusso NF, Lindor KD, Dickson ER. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc 2000;75(7):688-94. [ Links ]
16. Navaneethan U, Venkatesh PG, Lashner BA, Shen B, Kiran RP. The impact of ulcerative colitis on the long-term outcome of patients with primary sclerosing cholangitis. Aliment Pharmacol Ther 2012;35(9):1045-53. [ Links ]
17. Joo M, Abreu e Lima P, Farraye F, Smith T, Swaroop P, Gardner L, Lauwers GY, Odze RD. Pathologic features of ulcerative colitis in patients with primary sclerosing cholangitis: a case-control study. Am J Surg Pathol 2009;33(6):854-62. [ Links ]
18. Boonstra K, Van Erpecum KJ, Van Nieuwkerk KMJ, Drenth JP, Poen AC, Witteman BJ, Tuynman HA, Beuers U, Ponsioen CY. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm Bowel Dis 2012;18(12):2270-6. [ Links ]
19. Sano H, Nakazawa T, Ando T, Hayashi K, Naitoh I, Okumura F, Miyabe K, Yoshida M, Takahashi S, Ohara H, Joh T. Clinical characteristics of inflammatory bowel disease associated primary sclerosing cholangitis. J Hepatobiliary Pancreat sci 2011;18(2):154-61. [ Links ]
20. Sokol H, Cosnes J, Chazouilleres O, Beaugerie L, Tiret E, Poupon R, Seksik P. Disease activity and cancer risk in inflammatory bowel disease associated with primary sclerosing cholangitis. World J Gastroenterol 2008;14(22):3497-03. [ Links ]
21. Torres J, Pineton de Chambrun G, Itzkowitz S, Sachar DB, Colombel JF. Review article: colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease. Aliment Pharmacol Ther 2011;34(5):497-508. [ Links ]
22. Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. Gastrointest Endosc 2002;56(1):48-54. [ Links ]
23. Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology 1995;22(5):1404-8. [ Links ]
24. Moayyeri A, Daryani NE, Bahrami H, Haghpanah B, Nayyer-Habibi A, Sadatsafavi M. Clinical course of ulcerative colitis in patients with and without primary sclerosing cholangitis. J Gastroenterol Hepatol 2005;20(3):366-70. [ Links ]
25. Ngu JH, Gearry RB, Wright AJ, Stedman CA. Inflammatory bowel disease is associated with poor outcomes of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2011;9(12):1092-7. [ Links ]
26. Navaneethan U, Venkatesh PG, Mukewar S, Lashner BA, Remzi FH, Mccullough AJ, Kiran RP, Shen B, Fung JJ. Progressive primary sclerosing cholangitis requiring liver transplantation is associated with reduced need for colectomy in patients with ulcerative colitis. Clin Gastroenterol Hepatol 2012;10(5):540-6. [ Links ]
27. Marelli L, Xirouchakis E, Kalambokis G, Cholongitas E, Hamilton Mi, Burroughs AK. Does the severity of primary sclerosing cholangitis influence the clinical course of associated ulcerative colitis? Gut 2011;60(9):1224-8. [ Links ]
28. Vera A, Moledina S, Gunson B, Hubscher S, Mirza D, Olliff S, Neuberger J. Risk factors for recurrence of primary sclerosing cholangitis of liver allograft. Lancet 2002;360(9349):1943-4. [ Links ]
29. Cholongitas E, Shusang V, Papatheodoridis GV, Marelli L, Manousou P, Rolando N, Patch D, Rolles K, Davidson B, Burroughs AK. Risk factors for recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl 2008;14(2):138-43. [ Links ]
30. Alabraba E, Nightingale P, Gunson B, Hubscher S, Olliff S, Mirza D, Neuberger J. A re-evaluation of the risk factors for the recurrence of primary sclerosing cholangitis in liver allograft. Liver Transpl 2009;15(3):330-40. [ Links ]
31. Loftus EV JR., Aguilar Hi, Sandborn WJ, Tremaine WJ, Krom RA, Zinsmeister AR, Graziadei IW, Wiesner RH. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology 1998;27(3):685-90. [ Links ]
32. Jørgensen KK, Grzyb K, Lundin KE, Clausen OP, Aamodt G, Schrumpf E, Vatn MH, Boberg KM. Inflammatory bowel disease in patients with primary sclerosing cholangitis: clinical characterization in liver transplanted and nontransplanted patients. Inflamm Bowel dis 2012;18(3):536-45. [ Links ]
33. Gelley F, Miheller P, Péter A, Telkes G, Nemes B. Activity of ulcerative colitis before and after liver transplantation in primary sclerosing cholangitis: the Hungarian experience. Transplant Proc 2012;44(7):2164-5. [ Links ]
34. Penna C, Dozois R, Tremaine W, Sandborn W, Larusso N, Schleck C, Ilstrup D. Pouchitis after ileal pouch-anal anastomosis for ulcerative colitis occurs with increased frequency in patients with primary sclerosing cholangitis. Gut 1996;38(2):234-9. [ Links ]
35.Rahman M, Desmond P, Mortensen N, Chapman RW.The clinical impact of primary sclerosing cholangitis in patients with anileal pouch-anal anastomosis for ulcerative colitis. Int J Colorectal dis 2011;26(5):553-9. [ Links ]
36. Shen B, Bennett AE, Navaneethan U, Lian L, Shao Z, Kiran RP, Fazio VW, Remzi FH. Primary sclerosing cholangitis is associated with endoscopic and histological inflammation of the distal afferent limb in patients with ileal pouch-anal anastomosis. Inflamm Bowel dis 2011;17(9):1890-900. [ Links ]
37. Joshi D, Bjarnason I, Belgaumkar A, OGrady J, Suddle A, Heneghan MA, Aluvihare V, Rela M, Heaton N, Agarwal K. The impact of inflammatory bowel disease post-liver transplantation for primary sclerosing cholangitis. Liver int 2013;33(1):53-61. [ Links ]
38. Vera A, Gunson BK, Ussatoff V, Nightingale P, Candinas D, Radley S, Mayer A, Buckels JA, Mcmaster P, Neuberger J, Mirza DF. Colorectal cancer in patients with inflammatory bowel disease after liver transplantation for primary sclerosing cholangitis. Transplantation 2003;75(12):1983-8. [ Links ]
39. Van de Vrie W, De Man RA, Van Buuren Hr, Schouten Wr, Tilanus Hw, Metselaar HJ. Inflammatory bowel disease and liver transplantation for primary sclerosing cholangitis. Eur J Gastroenterol Hepatol 2003;15(6):657-63. [ Links ]
40. Dvorchik I, Subotin M, Demetris AJ, Fung JJ, Starzl Te, Wieand S, Abu-elmagd KM. Effect of liver transplantation on inflammatory bowel disease in patients with primary sclerosing cholangitis. Hepatology 2002;35(2):380-4. [ Links ]
41. Bergquist A, Glaumann H, Persson B, Broomé U. Risk factors and clinical presentation of hepatobiliary carcinoma in patients with primary sclerosing cholangitis: a case-controlstudy.Hepatology 1998;27(2):311-6. [ Links ]
42. Vierling JM. Aetiopathogenesis of primary sclerosing cholangitis. in: Manns P, Stieihl A, Wiesner R, editors. Primary sclerosing cholangitis. londres: Kluwer Academic Publishers;1998. p. 9. [ Links ]
43. Worthington J, Chapman R. Primary sclerosing cholangitis. Orphanet J Rare dis 2006;1:41. [ Links ]
44. Björnsson E, Cederborg A, Akvist A, Simren M, Stotzer Po, Bjarnason I. Intestinal permeability and bacterial growth of the small bowel in patients with primary sclerosing cholangitis. Scand J Gastroenterol 2005;40(9):1090-4. [ Links ]
45. Lichtman SN , Sartor RB. Hepatobiliary injury associated with experimental small-bowel bacterial overgrowth in rats. Immunol res 1991;10(3-4):528-31. [ Links ]
46. Grant AJ, Lalor Pf, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet 2002;359(9301):150-7. [ Links ]
47. Hillan KJ, Hagler Ke, Macsween RN, Ryan AM, Renz Me, Chiu HH, Ferrier RK, Bird Gl, Dhillon AP, Ferrell LD, Fong S. Expression of the mucosal vascular addressin, MAdCAM-1, in inflammatory liver disease. Liver 1999;19(6):509-18. [ Links ]
48. Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hübscher SG, Briskin M, Salmon M, Adams DH. Hepatic Endothelial CCL 25 mediates the recruitment of CCR 9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med 2004;200(11):1511-7. [ Links ]
49. Eksteen B, Mora JR, Haughton EL, Henderson NC, Lee-Turner L, Villablanca EJ, Curbishley SM, Aspinall AI, Von Andrian UH, Adams DH. Gut homing receptors on cd 8 T cells are retinoic acid dependent and not maintained by liver dendritic or stellate cells. Gastroenterol 2009;137(1):320-9. [ Links ]
50. Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol 2006;6(3):244-51. [ Links ]
51. Eksteen B, Miles AE, Grant AJ, Adams DH. Lymphocyte homing in the pathogenesis of extra-intestinal manifestations of inflammatory bowel disease. Clin Med 2004;4(2):173-80. [ Links ]
52. Fickert P, Moustafa T, Trauner M. Primary sclerosing cholangitis – the arteriosclerosis of the bile duct?. Lipids Health dis 2007;6:3. [ Links ]
53. Bouzid D, Haddouk S, Amouri A, Bem Hadj Hmida Y, Tahri N, Masmoudi H. Contribution of immunofluorescence to identification and characterization of antineutrophil cytoplasmic antibodies in inflammatory bowel disease. Indian J Gastroenterol 2011;30(5):229-32. [ Links ]
54. Bansi DS, Chapman RW , Fleming KA. Prevalence and diagnostic role of antineutrophil cytoplasmic antibodies in inflammatory bowel disease. Eur J Gastroenterol Hepatol 1996;8(9):881-5. [ Links ]
55. Dobric S, Popovic D, Nikolic M, Andrejevic S, Spuran M, Bonaci-Nikolic B. Anti-neutrophil cytoplasmic antibodies (Anc A) specific for one or several antigens: useful markers for subtypes of ulcerative colitis and associated primary sclerosing cholangitis. Clin Chem Lab Med 2011;50(3):503-9. [ Links ]
56. Stoffel MP, Csernok E, Herzberg C, Johnston T, Carroll SF, Gross WL. Anti-neutrophil cytoplasmic antibodies (Anc A) directed against bactericidal /permeability increasing protein (BPI): a new seromarker for inflammatory bowel disease and associated disorders. Clin Exp Immunol 1996;104(1):54-9. [ Links ]
57. Weiss J, Elsbach P, Olsson I, Odeberg H. Purification and characterization of a potent bactericidal and membrane active protein from the granules of human polymorphonuclear leukocytes. J Biol Chem 1978;253(8):2664-72. [ Links ]
58. Weiss J, Elsbach P, Shu C, Castillo J, Grinna L, Horwitz A, Theofan G. Human bactericidal/permeability-increasing protein and recombinant nH2-terminal fragment cause killing of serum resistant Gram-negative bacteria in whole blood and inhibit tumor necrosis factor release induced by the bacteria. J Clin Invest 1992;90(3):1122-30. [ Links ]
59. Cooper T, Savige J, Nassis L, Paspaliaris B, Neeson P, Neil J, Knight KR, Daskalakis M, Doery JC. Clinical associations and characterization of antineutrophil cytoplasmic antibodies directed against bactericidal/permeability-increasing protein and azurodicin. Rheumatol int 2000;19(4):129-36. [ Links ]
60. Roozendaal C, Van Milligen DE Wit AW, Haagsma EB, Horst G, Schwarze C, Peter HH, Kleibeuker JH, Tervaert JW, Limburg PC, Kallenberg CG. Antineutrophil cytoplasmic antibodies in primary sclerosing cholangitis: defined specificities may be associated with distinct clinical features. Am J Med 1998;105(5):393-9. [ Links ]
61. Schultz H, Weiss J, Carroll SF , Gross WL . The endotoxin-binding bactericidal/permeability-increasing protein (BPI): a target antigen of autoantibodies. J Leukoc Biol 2001;69(4):505-12. [ Links ]
62. Baveye S, Elass E, Mazurier J, Spik G, Legrand D. Lactoferrin: a multifunctional glycoprotein involved in the modulation of the inflammatory process. Clin Chem Lab Med 1999;37(3):281-6. [ Links ]
63. Muratori L, Muratori P, Zauli D, Grassi A, Pappas G, Rodrigo L, Cassani F. Antilactoferrin antibodies in autoimmune liver disease. Clin Exp Immunol 2001;124(3):470-3. [ Links ]
64. Hov JR, Boberg KM, Karlsen TH. Autoantibodies in primary sclerosing cholangitis. World J Gastroenterol 2008;14(24):3781-91. [ Links ]
65. Orth T, Kellner R, Diekmann O, Faust J, Meyer Zum Büschenfelde KH, Mayet WJ. Identification and characterization of autoantibodies against catalase and a-enolase in patients with primary sclerosing cholangitis. Clin Exp Immunol 1998;112(3):507-15. [ Links ]
66. Terjung B, Spengler U, Sauerbruch T, Worman HJ. Atipical p-Anc A in iBd and hepatobiliary disorders react with a 50-kilodalton nuclear envelope protein of neutrophils and myeloid cell lines. Gastroenterology 2000;119(2):310-22. [ Links ]
67. Terjung B, Muennich M, Gottwein J, Soehne J. Identification of myeloid-specific tubulina-beta isotype 5 as target antigen of antineutrophil cytoplasmic antibodies in autoimmune disease. Hepatology 2005;42(4 suppl 1):288A. [ Links ]
68. Terjung B, Söhne J, Lechtenberg B, Gottwein J, Muennich M, Herzog V, Mähler M, Sauerbruch T, Spengler U. P-Anc As in autoimmune liver disease recognize human beta-tubulin isotype 5 and cross-react with microbial protein ftsZ. Gut 2010;59(6):808-16. [ Links ]
69. Bi EF, Lutkenhaus J. ftsZ ring structure associated with division in escherichia coli. Nature 1991;354(6349):161-4. [ Links ]
70. Erickson HP. ftsZ, a prokaryotic homolog of tubulin?. Cell 1995;80(3):367-70. [ Links ]
71. Terjung B, Spengler U. Atypical p-Anc A in Psc and AiH: a hint toward a leaky gut?. Clin Rev Allergy immunol 2009;36(1):40-51. [ Links ]
72. Bergquist A, Montgomery SM, Bahmanyar S, Olsson R, Danielsson A, Lindgren S, Prytz H, Hultcrantz R, Lööf IA, Sandberg-Gertzén H, Almer S, Askling J, Ehlin A, Ekbom A. Increased risk of primary sclerosing cholangitis and ulcerative colitis in first-degree relatives of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2008;6(8):939-43. [ Links ]
73. Schrumpf E, Fausa O, Førre O, Dobloug JH, Ritland S, Thorsby E. HLA antigens and immunoregulatory T cells in ulcerative colitis associated with hepatobiliary disease. Scand J Gastroenterol 1982;17(2):187-91. [ Links ]
74. Karlsen TH, Franke A, Melum E, Kaser A, Hov Jr, Balschun T, Lie BA, Bergquist A, Schramm C, Weismüller TJ, Gotthardt D, Rust C,Philippe E, Fritz T, Henckaert Sl, Weersma RK, Stokkers P, Ponsioen CY, Wijmenga C, Sterneck M, Nothnagel M, Hampe J, Teufel A, Runz H, Rosenstiel P, Stiehl A, Vermeire S, Beuers U, Manns MP, Schrumpf E, Boberg KM, Schreiber S. Genome-Wide association analysis in primary sclerosing cholangitis. Gastroenterology 2010;138(3):1102-11. [ Links ]
75. Melum E, Franke A, Schramm C, Weismüller TJ, Gotthardt DN, Offner FA, Juran BD, Laerdahl JK, Labi V, Björnsson E, Weersma RK, Henckaerts l, Teufel A, Rust C, Ellinghaus E, Balschun T, Boberg KM, Ellinghaus D, Bergquist A, Sauer P, Ryu E, Hov Jr, Wedemeyer J, Lindkvist B, Wittig M, Porte RJ, Holm K, Gieger C, Wichmann HE, Stokkers P, Ponsioen CY, Runz H, Stiehl A, Wijmenga C,Sterneck M, Vermeire S, Beuers U, Villunger A, Schrumpf E, Lazaridis KN, Manns MP, Schreiber S, Karlsen TH. Genome-wide association analysis in primarysclerosingcholangitis identifies two non-HLA susceptibility loci. Nat Genet 2011;43(1):17-9. [ Links ]
76. Horton R, Wilming l, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC JR, Wright MW, Wain HM, Trowsdale J, Ziegler A, Beck S. Gene map of the extended human MHC. Nat Rev Genet 2004;5(12):889-99. [ Links ]
77. Janse M, Lamberts LE, Franke L, Raychaudhuri S, Ellinghaus E, Muri Boberg K, Melum E, Folseraas T, Schrumpf E, Bergquist A, Björnsson E, Fu J, Jan Westra H, Groen HJ, Fehrmann RS, Smolonska J, Van Den Berg lH, Ophoff RA, Porte RJ, Weismüller TJ, Wedemeyer J, Schramm C, Sterneck M, Günther R, Braun F, Vermeire S, Henckaerts L, Wijmenga C, Ponsioen CY, Schreiber S, Karlsen TH, Franke A, Weersma RK. Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, rel, and CARD9. Hepatology 2011;53(6):1977-85. [ Links ]
78. Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, Baldelli F, Donini A, Fiorucci S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. Plos One 2011;6(10):e25637. [ Links ]
79. Goyette P, Lefebvre C, Ng A, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Latiano A, Aumais G, Deslandres C, Jobin G, Annese V, Daly MJ, Xavier RJ, Rioux Jd. Gene-centric association mapping of chromosome 3p implicates MS T1 in IBd pathogenesis. Mucosal immunol 2008;1(2):131-8. [ Links ]
80.Hsu W, Zhang W,Tsuneyama K, Moritoki Y, Ridgway WM, Ansari AA, Coppel RL, Lian ZX, Macka YI, Gershwin ME. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus IBd in il -2ra -/-mice. Hepatology 2009;49(1):133-40. [ Links ]
81. Ruland J. CARD 9 signaling in the innate immune response. Ann N Y Acad SCI 2008;1143:35-44. [ Links ]
Gonçalo Figueiredo Esteves
Faculdade de Medicina da Universidade do Porto
Al. Prof. Hernâni Monteiro -4200-319 Porto
EMAIL: julio.p.costa@sapo.pt

