










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.29 no.1 Porto fev. 2015
ARTIGO DE REVISÃO
Doenças da comunicação intergenómica: abordagem clínica e laboratorial
Nuclear-mitochondrial intergenomic communication disorders: clinical and laboratory approach
Célia Nogueira1#, Ligia S Almeida1#, Arnaldo Videira2, Filippo M Santorelli3, Laura Vilarinho1
1Instituto Nacional de Saúde Doutor Ricardo Jorge, Departamento de Genética Humana – Unidade de Investigação & desenvolvimento, Porto
2ICBas - Instituto de Ciências Biomédicas de Abel Salazar, Universidade do Porto, Porto
3IRCCS Stella MarIs, molecular Medicine for Neuromuscular and Neurodegenerative diseases, Pisa, Itália
# Igual contribuição para este trabalho
RESUMO
As citopatias mitocondriais constituem um importante grupo de doenças metabólicas de expressão clínica heterogénea, para as quais não existe uma terapia eficaz. A maioria das doenças mitocondriais descritas é causada por uma disfunção ao nível do sistema da fosforilação oxidativa, originando consequentemente uma deficiente produção de energia. O correto funcionamento deste sistema resulta de uma interação coordenada entre os genomas nuclear e mitocondrial. Um defeito nesta interação origina as doenças da comunicação intergenómica, que causam a perda ou instabilidade do DNA mitocondrial (mtDNA) originando quer depleção quer deleções múltiplas neste genoma. Estas doenças adquiriram bastante relevância nos últimos anos, e podem ser classificadas em dois grandes grupos: i) síndrome das deleções múltiplas do mtDNA, que se caracteriza pelo aparecimento em adultos, de miopatias oculares e dos membros e ii) síndrome da depleção do mtDNA, que se manifesta durante a infância ou início da adolescência com três apresentações clínicas distintas, nomeadamente miopática, encefalomiopática e hepatocerebral. Neste trabalho pretendeu-se realizar uma revisão bibliográfica no sentido de apurar o estado da arte em relação às doenças da comunicação intergenómica. Deste modo, serão apresentados os fenótipos clínicos e o espetro mutacional associado, assim como um algoritmo de diagnóstico, que possa ser uma ferramenta útil para os clínicos que se dedicam ao diagnóstico e follow-up deste tipo de patologias.
Palavras-chave: depleção do mtDNA, deleções múltiplas do mtDNA, doenças mitocondriais, doenças da comunicação intergenómica
ABSTRACT
Mitochondrial dysfunction accounts for an important and heterogeneous group of inherited metabolic disorders with hitherto no effective therapeutic options. most of the known mitochondrial disorders are caused primarily by a dysfunctional oxidative phosphorylation and consequently a deficient energy production. This system depends on the coordinated expression of both nuclear and mitochondrial genomes. Therefore, mitochondrial diseases can be caused by genetic defects in the mitochondrial or the nuclear genome or in the interplay between the two genomes, causing nuclear-mitochondrial intergenomic communication disorders. The mitonuclear crosstalk has gained increased relevance in the past years and since then many genes have been identified as being involved in these diseases. Nuclear-mitochondrial intergenomic communication disorders can be divided into two distinct groups: i) multiple deletions syndrome, leading to qualitative changes of mtDNA, that occurs in adults, having as main clinical features ocular and limb myopathy, which are almost always associated with extramuscular system involvement or ii) depletion syndrome, leading to quantitative changes of mtDNA, occurring in infancy or early childhood further subdivided into three clinical categories: myopathic, encephalomyopathic and hepatocerebral. The focus of this review is to provide an overview of intergenomic communication disorders, a list of clinical phenotypes accompanied by their mutational spectrum and a diagnostic algorithm, which can be useful for clinicians when facing similar cases.
Key-words: mitochondrial DNA depletion, mitochondrial DNA multiple deletions, mitochondrial diseases, intergenomic communication disorders
Introdução
Nos últimos 30 anos, um largo espectro de doenças multissistémicas associadas a disfunções da mitocôndria, globalmente designadas de citopatias mitocondriais, têm sido descritas em doentes de diferentes grupos etários.1 Estas disfunções podem afetar qualquer órgão ou tecido do organismo, embora os músculos esquelético e cardíaco, e o sistema nervoso central sejam os mais afetados, devido à sua elevada dependência do metabolismo energético. Assim se compreende que as formas de apresentação mais frequentes incluam miopatia, encefalo(mio)patia e oftalmoplegia externa progressiva (PEO). Aproximadamente 1:5.000 indivíduos na população adulta e infantil possuem citopatias mitocondriais ou correm o risco de as virem a desenvolver.2 Estas doenças são uma causa comum de mortalidade e/ou morbilidade crónica, não estando disponível, salvo raras exceções, nenhuma terapia eficaz. Desde1988, que a investigação molecular das citopatias mitocondriais tem sido focada no DNA mitocondrial (mtDNA) nomeadamente na pesquisa de rearranjos do mtDNA (deleções simples de diferentes tamanhos e duplicações) e pesquisa de mutações pontuais quer nos genes estruturais quer nos 22 tRNAs e dois rRNAs.3 Neste sentido, para aprofundar o conhecimento das citopatias mitocondriais em Portugal, implementou-se em 1993, na nossa Unidade, o estudo enzimático da cadeia respiratória mitocondrial e posteriormente o estudo molecular do mtDNA.4,5,6,7
Nos últimos anos a investigação destas doenças tem-se centrado na pesquisa de mutações nos genes nucleares que codificam subunidades estruturais da cadeia respiratória mitocondrial assim como nos genes necessários à montagem das mesmas ou envolvidos na comunicação mito-nuclear.
Neste trabalho efetuou-se uma revisão da literatura sobre as doenças da comunicação intergenómica, focando os principais grupos de patologias, fenótipos clínicos e respetivo espetro mutacional.
Manifestações clínicas e etiologia molecular das patologias que afetam a integridade do mtDNA
A integridade do mtDNA é mantida e controlada por um mecanismo complexo que envolve vários elementos do replissoma mitocondrial e diversas enzimas e transportadores que fornecem à mitocôndria os nucleótidos necessários (Fig. 1). todos estes componentes são codificados pelo DNA nuclear (nDNA) assim mutações nos genes envolvidos na replicação e manutenção do mtDNA podem comprometer a sua integridade causando quer deleções múltiplas quer depleção do mtDNA.8,9 Nos últimos anos a comunicação entre os dois genomas tem ganho particular importância e muitos genes têm sido identificados como estando envolvidos neste grupo de citopatias mitocondriais.
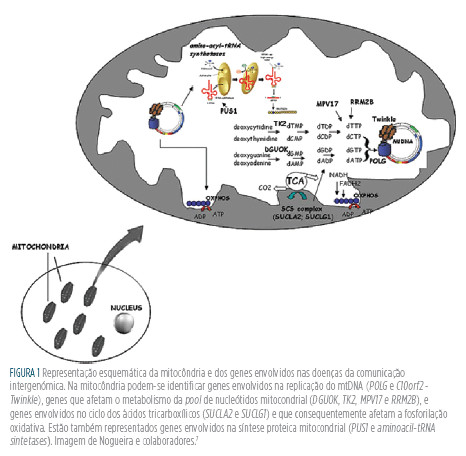
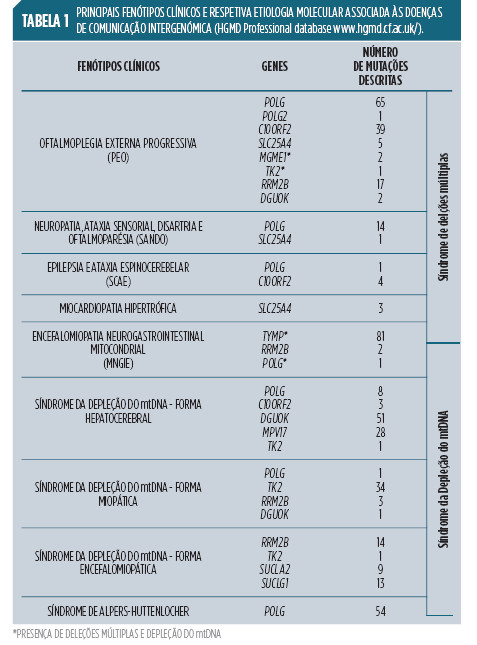
A Tabela 1
sumariza as principais manifestações clínicas associadas à síndrome das deleções múltiplas e à da depleção do mtDNA, assim como a respetiva etiologia molecular.
1. Síndrome das deleções múltiplas do mtDNA
As doenças mitocondriais associadas à presença de deleções múltiplas do mtDNA ocorrem mais frequentemente em adultos e podem apresentar, na sua maioria, uma hereditariedade autossómica recessiva ou dominante, sendo esta última mais frequente.10 O tamanho das diferentes deleções do mtDNA é variável, até mesmo em doentes da mesma família. O espetro mutacional desta síndrome tem vindo a aumentar devido à descoberta de cada vez mais genes associados à instabilidade do mtDNA. Seguidamente será efetuada uma breve descrição das principais manifestações clínicas e asetiologias moleculares associadas.
i) Peo (Oftalmoplegia externa progressiva autossómica dominante ou recessiva)
As características clínicas mais comuns da PEO no adulto incluem a fraqueza dos músculos oculares externos, ptose bilateral, fraqueza muscular proximal e intolerância ao exercício. Sintomas como a presença de cataratas, perda de audição, neuropatia axonal sensorial, ataxia, depressão, hipogonadismo, e parkinsonismo também podem ocorrer. Características menos comuns incluem prolapso da válvula mitral, miocardiopatia e alterações da motilidade gastrointestinal. Os indivíduos afetados apresentam deleções múltiplas ao nível do mtDNA que se encontram exclusivamente nos tecidos musculares.
Mutações nos genes POLG, POLG2, C10orf2, SLC25A4, TK2, RRM2B, DGUOK, MGME1 OPA1 e MFN2, encontram-se associados a PEO podendo apresentar uma hereditariedade autossómica recessiva ou dominante sendo a forma autossómica recessiva geralmente mais grave.10,11
Seguidamente são referidos os principais genes envolvidos na etiologia molecular de PEO associados à síndrome das deleções múltiplas.
POLG: a mitocôndria contêm uma única DNA polimerase, a polimerase gama (POlγ), imprescindível desde muito cedo na embriogénese.12 A POlγ é codificada pelo nDNA sendo a única polimerase exclusivamente responsável pela replicação e reparação do mtDNA e essencial na sua manutenção. Esta enzima é composta por uma subunidade catalítica, POlγA, que possui atividade de polimerase e de exonuclease, sendo codificada pelo gene POLG; e uma subunidade acessória, POlγB, que potencia a ação da enzima.13 A POlγ juntamente com o mtDNA, a helicase do mtDNA e as diversas proteínas de ligação formam o complexo de replicação funcionante.14
As mutações que se encontram no gene POLG são uma das principais causas da doença mitocondrial podendo estar associadas a uma grande variedade de sintomas clínicos, para além de PEO, tais como: parkinsonismo, síndrome de Alpers -Huttenlocher, encefalomiopatia neurogastrointestinal mitocondrial (MNGIE), neuropatia ataxia sensorial disartria e oftalmoparesia (SANDO) e ataxia espinocerebelar e epilepsia (SCAE).15,16,17
Este gene está localizado no cromossoma 15 e é composto por 23 exões que se estendem ao longo de 18,55 Kb. Apesar de ter sido identificado em 199618, somente em 2001 foi descrita a primeira mutação patogénica.19 Até à data, mais de 200 mutações foram publicadas, das quais 65 foram descritas associadas a PEO, sendo este gene um hot-spot para mutações em doenças mitocondriais (human gene mutation Database-HGMD, POLG database).20 Mutações no POLG podem também ser associadas à forma hepatocerebral da síndrome da depleção do mtDNA.15
Mutações neste gene podem apresentar uma hereditariedade autossómica recessiva ou autossómica dominante ou até a mesma mutação pode apresentar ambos os padrões de hereditariedade.21,22
POLG2: a POlγB, subunidade acessória da POl?, codificada pelo gene POLG2, aumenta a eficiência da enzima. Alterações nesta subunidade podem causar deleções múltiplas do mtDNA.
O gene POLG2, localizado no cromossoma 17, é composto por oito exões que se distribuem ao longo de 19,28 Kb. A primeira mutação patogénica foi descrita em 2006 associada à forma autossómica dominante de PEO.23 posteriormente mais 12 mutações foram descritas neste gene associadas a fenótipos mitocondriais inespecíficos (HGMD).
C10orf2 (Twinkle): a helicase/primase mitocondrial codificada pelo gene C10orf2 também é responsável pela forma autossómica dominante de PEO.24 mutações neste gene estão associadas a apresentações clínicas que podem apresentar PEO puro, ou uma forma mais complexa de PEO associada a fraqueza muscular proximal dos membros e músculos faciais, disfagia e disfonia, ataxia, e neuropatia periférica.
O gene C10orf2 está localizado no cromossoma 10, sendo composto por 5 exões ao longo de 6,38 kb. A primeira mutação patogénica, associada a PEO, foi descrita em 200124 e desde então mais de 40 mutações patogénicas foram publicadas (HGMD). Algumas mutações neste gene foram também identificadas na forma hepatocerebral da síndrome da depleção do mtDNA.25,26
SLC25A4 (ANT1): este gene codifica o transportador mitocondrial de adenina específico do músculo cardíaco (ANT1). A proteína ANT1 é a proteína mitocondrial mais abundante e, no seu estado funcional, é um homodímero de subunidades de 30 kD incorporados assimetricamente na membrana mitocondrial interna. O homodímero forma um poro através do qual o ADP é transportado da matriz para o citoplasma. Nos mamíferos podem distinguir-se quatro isoformas específicas em diferentes tecidos.
O gene SLC25A4 está localizado no cromossoma 4 e possui 4 exões distribuídos ao longo de 4,04 Kb. A primeira mutação patogénica foi descrita em 200027 e, desde então, apenas nove mutações estão referenciadas. Destas, cinco estão associadas à forma autossómica dominante de PEO, três estão associadas a miopatia e a miocardiopatia hipertrófica e uma foi identificada num doente com SANDO.
MGME1: o gene MGME1 foi recentemente descrito28 e localiza-se no cromossoma 20. É composto por quatro exões e codifica uma exonuclease mitocondrial que é essencial para uma síntese eficaz do mtDNA. É a primeira exonuclease mitocondrial identificada a estar envolvida na replicação do mtDNA, podendo ter uma função adicional de reparação, através do reposicionamento da cadeia de DNA ou das estruturas de DNA-rNA durante a síntese do mtDNA.
Encontram-se descritas duas mutações em três famílias distintas com PEO, que apresentam perda de peso acentuada e insuficiência respiratória.28 As biópsias musculares destes doentes revelaram quer deleções múltiplas, quer depleção do mtDNA.
ii) SANDO (NeuropatIa, ataxia sensorIal, disartria e oftalmoparesia)
SANDO é uma doença sistémica, autossómica recessiva, caracterizada principalmente pelo aparecimento de neuropatia, ataxia sensorial, disartria e oftalmoparésia em adultos. O fenótipo é muito variável, mesmo dentro da mesma família, e pode ainda incluir miopatia, convulsões e perda de audição.29 Até à data, foram descritas 14 mutações no gene POLG associadas a este fenótipo (HGMD).16
iii) SCAE (Epilepsia e ataxia espinocerebelar)
SCAE é uma patologia semelhante a SANDO mas com uma maior frequência de enxaquecas e convulsões.30 Estão descritas quatro mutações no gene C10orf2 e uma no gene POLG associadas a esta doença.
iv) MNGIE (Encefalomiopatia neurogastrointestinal mitocondrial)
MNGIE é uma doença autossómica recessiva com início entre a segunda e quinta décadas de vida caracterizada clinicamente por PEO, (pseudo) dismotilidade gastrointestinal, caquexia, leucoencefalopatia difusa, neuropatia periférica e morte precoce. Ao nível do mtDNA podem ser observadas deleções múltiplas e/ou depleção do mtDNA.31 mutações nos genes TYMP e RRM2B e mais recentemente no gene POLG32 foram relacionadas com este fenótipo. Seguidamente é referido o principal gene envolvido na etiologia molecular de MNGIE.
TYMP (ECGF1): o gene TYMP codifica a enzima timidina fosforilase (TP), que está envolvida no catabolismo das pirimidinas. Défices da enzima tp levam a uma acumulação sistémica de timidina e deoxiuridina, o que leva a um desequilíbrio da pool de deoxinucleótidos e consequentemente a uma instabilidade do mtDNA, originando o aparecimento quer de deleções múltiplas quer de depleção do mtDNA.33
O gene TYMP está localizado no cromossoma 22 e é composto por 10 exões que se estendem ao longo de 4,3 kb. As primeiras mutações patogénicas foram descritas por Nishino e colaboradores33 e, desde então, 80 mutações associadas a este fenótipo foram publicadas neste gene.
2. Síndrome da depleção do mtDNA
A síndrome da depleção do mtDNA caracteriza-se por uma redução acentuada do número de cópias do mtDNA em relação às do nDNA, associada muitas vezes a um défice múltiplo dos complexos enzimáticos da cadeia respiratória mitocondrial. Este grupo de doenças raras e devastadoras manifesta-se maioritariamente logo após o nascimento, causando a morte prematura de muitos doentes durante a infância ou início da adolescência. Esta síndrome é transmitida de modo autossómico recessivo e pode apresentar-se sob três formas: miopática, encefalomiopática e hepatocerebral. Embora a maioria destas manifestações ocorram em tecidos específicos, afetando fundamentalmente o fígado, o músculo ou o cérebro, outros tecidos podem também ficar comprometidos na evolução da doença.34 O seu mecanismo fisiopatológico está fundamentalmente relacionado com a manutenção da pool de nucleótidos mitocondriais, que assume um papel crucial na replicação e integridade do mtDNA.
A depleção do mtDNA (número de cópias do mtDNA inferior a 35%) nos tecidos afetados, con-firma o diagnóstico clínico desta síndrome.
Seguidamente é efetuada uma breve descrição das principais formas da síndrome de depleção do mtDNA, assim como dos genes envolvidos na sua etiologia molecular.
i) Forma hepatocerebral
A forma hepatocerebral é a apresentação mais comum desta síndrome. Os primeiros sintomas ocorrem entre o nascimento e os 6 meses de vida e incluem vómitos persistentes, atraso de crescimento, hipotonia e hipoglicemia. A nível histológico pode-se encontrar esteatose microvacuolar, colestase, fibrose e cirrose. Estes doentes têm normalmente uma morte precoce, durante o primeiro ano de vida.Uma apresentação peculiar da forma hepatocerebral é a síndromede Alpers-Huttenlocher, caracterizada por falência hepática, convulsões, evoluindo para epilepsia parcial contínua e deterioração neurológica global. O uso de ácido valpróico como tratamento para a epilepsia pode precipitar a falência hepática aguda. Mutações no gene POLG são uma causa frequente desta síndrome. Até ao momento, estão descritas cerca de 55 mutações associadas à forma hepatocerebral, distribuídas pelos seguintes genes: DGUOK, MPV17, POLG, C10orf2, TK2 e SUCLG1. Genes, como DGUOK e MPV17, encontram-se também associados à síndrome das deleções múltiplas do mtDNA.35,36 Uma breve descrição destes genes será efetuada seguidamente, com exceção dos genes POLG e C10orf2, que foram descritos na secção anterior e dos genes TK2 e SUCLG1que serão descritos posteriormente nas formas miopática e encefalomiopática desta síndrome, respetivamente.
DGUOK: a cínase deoxiguanosina (DGUOK) fosforila a deoxiguanosina e a deoxiadenosina, contribuindo para a síntese de dois nucleótidos necessários à manutenção da pool de desoxirribonucleótidos trifosfato (dNTPs) mitocondrial.22,37 A associação da síndrome da depleção do mtDNA com mutações neste gene sugere que a manutenção desta pool de dNTPs representa um papel crucial na replicação e integridade do mtDNA e consequentemente no seu conteúdo. Os doentes com mutações no gene DGUOK, localizado no locus 2p13, apresentam geralmente alterações hepáticas progressivas e dificuldades alimentares, associadas a disfunção neurológica (hipotonia, nistagmus e atraso psicomotor) por volta dos primeiros meses de vida. A presença de neuropatia periférica e tubulopatia são raras mas, foram descritas ocasionalmente.38 mutações neste gene causam depleção do mtDNA fundamentalmente no fígado, e mais raramente no músculo e nos fibroblastos. As primeiras mutações patogénicas descritas neste gene foram publicadas em 2001 por Mandel e colaboradores.39 Atualmente, foram descritas mais de 50 mutações em cerca de 100 doentes (HGMD).
MPV17: outro gene envolvido nesta forma clínica é o MPV17 que se localiza no locus 2p23-p21 e codifica uma pequena proteína da membrana mitocondrial interna de função ainda pouco esclarecida. Estima-se que participa na manutenção da pool de dNTPs necessária para a síntese de mtDNA. O seu papel na patogénese e desta síndrome é ainda desconhecido. Clinicamente estes doentes apresentam falência hepática grave, hipoglicemia, sintomas neurológicos e múltiplas lesões a nível cerebral durante o primeiro ano de vida.40 A presença de depleção do mtDNA no fígado e/ou no músculo, assim como um défice múltiplo dos complexos da cadeia respiratória mitocondrial, estão normalmente presentes nestes doentes. Foram descritos aproximadamente 30 indivíduos com mutações no gene MPV17.41,42 Desde a identificação da primeira mutação neste gene43, foram descritas mais de 25 mutações associadas a esta forma clínica e também à neuro-hepatopatia de Navajo, uma doença multissistémica autossómica recessiva encontrada na comunidade de Navajo no sudoeste dos Estados Unidos.22
ii) Forma miopática
Na forma miopática da síndrome da depleção do mtDNA, o aparecimento de sintomas ocorre normalmente no primeiro ano de vida, com dificuldades alimentares, atraso de crescimento, hipotonia e fraqueza muscular. A creatina cínase está frequentemente aumentada, sendo um marcador importante para o diagnóstico, uma vez que não é muito comum em doentes com outras miopatias mitocondriais.9 A morte destes doentes ocorre normalmente nos primeiros anos de vida devido a insuficiência pulmonar e infeções, mas existem casos que sobreviveram até à adolescência.44 Estas manifestações clínicas e bioquímicas são acompanhadas de sinais morfológicos típicos de miopatia mitocondrial tal como a presença de fibras citocromo c oxidase negativas. A proliferação de mitocôndrias sob a forma de fibras rotas e vermelhas não é uma característica primária consistente mas, no entanto, pode aparecer mais tarde no decorrer da doença.
TK2: a timidina cínase (TK2) é uma enzima que fosforila a deoxitimidina, deoxicitosina e deoxiuridina, participando juntamente com a DGUOK na síntese de nucleótidos necessários para a manutenção da pool de dNTPs mitocondrial.45
O gene TK2 localiza-se no locus 16q22 e as mutações descritas neste gene estão especificamente associadas à forma miopática. Os sintomas aparecem durante a infância ou a adolescência e caracterizam-se por miopatia progressiva, culminando em falência respiratória e em morte, nos casos mais graves. No entanto, os fenótipos menos graves apresentam uma evolução mais lenta da doença e um maior tempo de sobrevivência.34
Em 2001, Saada e colaboradores46 identificaram pela primeira vez mutações neste gene. Depois desta primeira observação, um total de 37 mutações no TK2 foram já descritas em mais de 50 doentes.42 A maioria destas mutações estão associadas à forma miopática. No entanto, existem mutações associadas a outros fenótipos, como é o caso da PEO, surdez neurosensorial e encefalomipatia com epilepsia (HGMD).
iii) Forma encefalomiopática
Em 2005, Elpeleg e colaboradores47, descreveram uma forma de encefalomiopatia autossómica recessiva associada a depleção do mtDNA. O aparecimento dos sintomas ocorre igualmente na infância e manifesta-se através de hipotonia muscular, atraso psicomotor grave, deterioração neurológica progressiva, surdez, perda de movimentos voluntários, oftalmoplegia externa, convulsões generalizadas e disfunção tubular renal. Verifica-se um aumento do lactato sanguíneo e na ressonância magnética cerebral, podem-se observar lesões nos gânglios basais sugestivas da síndrome de leigh.37
Mutações nos genes, RRM2B, SUCLA2 e SUCLG1 estão associadas a esta forma de apresentação clínica, sendo estes seguidamente descritos.
RRM2B: o gene RRM2B localiza-se no locus8q23 e codifica a subunidade R2 da redutase ribonucleotídica (RNR), uma enzima citosólica que está envolvida no passo terminal da síntese de novo dos dNTPs, convertendo os nucleótidos em dNTPs durante a interfase do ciclo celular (fase S). A RNR está também envolvida no suplemento de dNTPs durante a reparação do DNA. Mutações neste gene estão associadas a hipotonia, acidose láctica, atraso de desenvolvimento e tubulopatia. A doença apresenta uma evolução rápida e fatal e manifesta-se frequentemente nos primeiros meses de vida. Laboratorialmente é identificada uma diminuição acentuada do número de cópias de mtDNA no músculo destes doentes.
Em 2007, Bourdon e colaboradores48 descreveram a primeira mutação patogénica neste gene, estando atualmente mais de 30 mutações descritas em aproximadamente 15 doentes.42
SUCLA2 e SUCLG1: a enzima succinil-coA sintetase catalisa a síntese reversível do succinato e ATP a partir de succinil-coA e ADP no ciclo dos ácidos tricarboxílicos (TCA). Esta enzima é constituída por duas subunidades, a e ß, codificadas pelos genes SUCLG1 (2p11) e SUCLA2 (13q12), respetivamente. Mutações nestes genes parecem dificultar a associação entrea succinil-coA sintetase e a cínasedifosfato nucleósido, resultando num desequilíbrio na pool de dNTPs mitocondrial e consequentemente na depleção do mtDNA no músculo.49 Um dado importante para a suspeita do envolvimento destes genes é a presença de uma excreção urinária, moderada a elevada, de ácido metilmalónico e a presença de intermediários do TCA. Clinicamente revelam hipotonia muscular proeminente, atraso psicomotor grave, distonia progressiva, surdez e convulsões generalizadas. Os doentes com mutações no gene SUCLG1 podem ainda apresentar dismorfias pré-natais, crises metabólicas neonatais e a morte pode ocorrer nos primeiros meses de vida.
Estão descritos cerca de 30 doentes com mutações nestes genes, das quais nove no SUCLA250 e 13 no SUCLG1 (HGMD).
Abordagens de diagnóstico das doenças de comunicação intergenómica
Nas doenças da comunicação intergenómica, a apresentação clínica pode variar entre as síndromes bem definidas e os fenótipos multissistémicos inespecíficos, onde o envolvimento neurológico está geralmente presente. Estabelecer um diagnóstico preciso num doente com suspeita de patologia mitocondrial revela-se um desafio, que requer uma abordagem multidisciplinar a nível clínico, bioquímico e histopatológico, para além de uma história familiar bem documentada. Os dados bioquímicos, tais como aumento de lactato e piruvato, quer no plasma, no LCR ou na urina, alteração do perfil de ácidos orgânicos, aumento da alanina no perfil dos aminoácidos plasmáticos, assim como os dados de ressonância/tomografia cerebral são indicadores importantes para o diagnóstico destas patologias. A determinação da atividade enzimática dos complexos enzimáticos da cadeia respiratória mitocondrial é importante para orientar a abordagem molecular, em particular em doentes sem fenótipo específico. Como o mtDNA codifica subunidades de vários complexos da fosforilação oxidativa (CI, CIII, CIV e CV), é expetável que uma depleção do mtDNA provoque um défice combinado da fosforilação oxidativa (com exceção do CII, o único não codificado pelo mtDNA). No entanto a atividade enzimática dos complexos da cadeia respiratória mitocondrial no músculo pode ser normal, se o tecido muscular não se encontrar entre os tecidos afetados, como acontece nos doentes com formas hepatocerebrais da síndrome da depleção do mtDNA.
A análise por southern-blot ou por PCR quantitativo em tempo real identifica simultaneamente as deleções múltiplas e a depleção do mtDNA. Independentemente da tecnologia aplicada utiliza-se um gene nuclear de referência específico para a análise (18s) sendo muito importante o uso de controlos da mesma idade e do mesmo tecido, dada a natureza dinâmica da qualidade e quantidade do mtDNA.51 considera-se como cut-off para o diagnóstico de depleção do mtDNA um número de cópias de mtDNA inferior a 35% em relação aos indivíduos controlo, podendo este número atingir valores mais baixos nos casos mais graves.
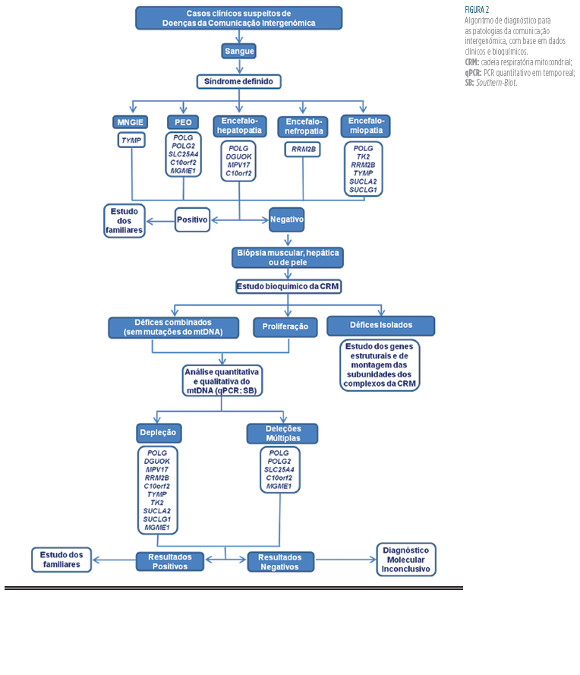
Com base na nossa experiência e na revisão da literatura publicada, apresentamos um algoritmo para o estabelecimento de um diagnóstico para estas doenças da comunicação intergenómica (Fig. 2
).
Considerações finais
As citopatias mitocondriais não dispõem de terapia eficaz,àexceção dosdéfices primários de ubiquinona (CoQ10) que respondem clinicamente à suplementação com CoQ10, embora sejam muito raros. Em geral, o tratamento efetuado nestas doenças é meramente sintomático e paliativo, usando-se cocktails vitamínicos, cofatores e eventualmente creatina e carnitina.
A investigação em termos de diagnóstico neste grupo de patologias não é muito diferente da que se aplica noutras doenças e inclui a recolha de dados como história familiar, exame físico incluindo o neurológico, exames laboratoriais de rotina e específico se eventual realização de biópsia muscular para estudo histopatológico, bioquímico e molecular.52 Uma doença mitocondrial que se manifesta ao nascimento ou nos primeiros dias de vida é mais provável estar associada com alterações a nível do nDNA do que com alterações a nível do mtDNA. As síndromes da depleção e das deleções múltiplas do mtDNA têm surgido cada vez mais como uma das principais causas de um amplo espetro de patologias multissistémicas de início na infância ou na idade adulta, respetivamente. Os avanços recentes na tecnologia de sequenciação, nomeadamente a utilização da sequenciação de nova geração, permitiram esclarecer a etiologia molecular em 55% dos doentes com suspeita de patologia mitocondrial, possibilitando a identificação de mutações causais em novos genes, por vezes não diretamente relacionados com patologias mitocondriais.53,54,55
A caraterização molecular destes doentes é importante não só para permitir a realização de aconselhamento genético e diagnóstico pré-natal adequados, nos casos em que forem identificadas mutações no nDNA, mas também para melhorar a compreensão da fisiopatologia da doença. Uma estratégia de diagnóstico precisa e focada irá economizar recursos e possibilitará avanços consideráveis no conhecimento destas patologias.
Agradecimentos
Lígia S Almeida tem financiamento da Fundação para a Ciência e Tecnologia ao abrigo do Programa Ciência 2008 (FCTC2008/INSA/ P4). Célia Nogueira teve financiamento da Fundação para a Ciência e Tecnologia (SFRH/BD/45247/2008).
Referências
1. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: role of inherited and somatic mutations. Nat Rev Genet 2012;13(12):879-90. [ Links ]
2. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders-past, present and future. Biochim Biophys Acta 2004;1659(2-3):115-20. [ Links ]
3. Wallace DC, Singh G, Lott MT , Hodge JA, Schurr TG , Lezza AM, Elsas LJ 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with Lebers hereditary optic neuropathy. Science 1988;242(4884):1427-30. [ Links ]
4. Vilarinho L, Maia C, Coelho T, Coutinho P, Santorelli FM. Heterogenous presentation in Leigh syndrome. J Inherit Metab Dis 1997;20(5):704-5. [ Links ]
5. Vilarinho L, Santorelli FM, Cardoso ML , Coelho T, Guimarães A, Coutinho P. Mitochondrial DNA analysis in ocular myopathy. Observations in 29 Portuguese patients. Eur Neurol 1998;39(3):148-53. [ Links ]
6. Vilarinho L, Leão E, Barbot C, Santos M, Rocha H, Santorelli FM. Clinical and molecular studies in three Portuguese mtDNA T8993G families. Pediatr Neurol 2000;22(1):29-32. [ Links ]
7. Nogueira C, Carrozzo R, Vilarinho L, Santorelli FM. Infantile-onset disorders of mitochondrial replication and protein synthesis. J Child Neurol 2011;26(7):866-75. [ Links ]
8. Hirano M, Marti R, Ferreiro-Barros C, Vilà MR , Tadesse S, Nishigaki Y, Nishino I, Vu TH . Defects of intergenomic communication: autosomal disorders that cause multiple deletions and depletion of mitochondrial DNA. Semin Cell Dev Biol 2001;12(6):417-27. [ Links ]
9. Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic signaling. Gene 2005;354(18):162-8. [ Links ]
10. Filosto M, Mancuso M, Nishigaki Y, Pancrudo J, Harati Y, Gooch C, Mankodi A, Bayne L, Bonilla E, Shanske S, Hirano M, DiMauro S. Clinical and genetic heterogeneity in progressive external ophthalmoplegia due to mutations in polymerase-gamma. Arch Neurol 2003;60(9):1279-84. [ Links ]
11. Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM , Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase-gamma mutations: clinical and molecular genetic study. Lancet 2004;364(9437):875-82. [ Links ]
12. Hance N, Ekstrand MI , Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet 2005;14(13):1775-83. [ Links ]
13. Lee Y-S, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009;139:312-24. [ Links ]
14. Korhonen JA, Pham XH, Pellegrini M, Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. EMBOJ 2004;23(12):2423-9. [ Links ]
15. Horvath R, Hudson G, Ferrari G, Fütterer N, Ahola S, Lamantea E, Prokisch H, Lochmüller H, McFarland R, Ramesh V, Klopstock T, Freisinger P, Salvi F, Mayr JA, Santer R, Tesarova M, Zeman J, Udd B, Taylor RW, Turnbull D, Hanna M, Fialho D, Suomalainen A, Zeviani M, Chinnery PF. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 2006;129(Pt 7):1674-84. [ Links ]
16. Gago MF, Rosas MJ, Guimarães J, Ferreira M, Vilarinho L, Castro L, Carpenter S. SANDO: two novel mutations in POLG 1 gene. Neuromuscul Disord 2006;16(8):507-9. [ Links ]
17. Ferreira M, Evangelista T, Almeida LS , Martins J, Macario MC , Martins E, Moleirinho A, Azevedo L, Vilarinho L, Santorelli FM. Relative frequency of known causes of multiple mtDNA deletions: two novel POLG mutations. Neuromuscul Disord 2011;21(7):483-8. [ Links ]
18. Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics 1996;36(3):449-58. [ Links ]
19. Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 2001;28(3):211-2. [ Links ]
20. Chinnery PF, Zeviani M. 155th ENMC workshop: polymerase gamma and disorders of mitochondrial DNA synthesis. Neuromuscul Disord 2008;18(3):259-67. [ Links ]
21. Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, Comi GP , Zeviani M. Mutations of mitochondrial DNA polymerase gamma are a frequent cause of autosomal dominant or recessive progressive external ophtalmoplegia. Ann Neurol 2002;52(2):211-9. [ Links ]
22. Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol 2012;47(1):64-74. [ Links ]
23. Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland, WC, Chinnery PF. Mutant POLG 2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet 2006;78(6):1026-34. [ Links ]
24. Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM , Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT , Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet 2001;28(3):223-31. [ Links ]
25. Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 2007;130(Pt 11):3032-40. [ Links ]
26. Nogueira C, Marques JS, Nesti C, Azevedo L, Di Lullo M, Meschini MC , Orlacchio A, Santorelli FM, Vilarinho L. Identification of maternal uniparental isodisomy of chromosome 10 in a patient with mitochondrial DNA depletion syndrome. Mol Genet Metab 2013;110(4):493-4. [ Links ]
27. Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP , Keranen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000;289(5480):782-5. [ Links ]
28. Kornblum C, Nicholls TJ, Haack TB, Schöler S, Peeva V, Danhauser K, Hallmann K, Zsurka G, Rorbach J, Iuso A, Wieland T, Sciacco M, Ronchi D, Comi GP , Moggio M, Quinzii CM , Dimauro S, Calvo SE, Mootha VK, Klopstock T, Strom TM , Meitinger T, Minczuk M, Kunz WS, Prokisch H. Loss-of-function mutations in MGM E1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet 2013;45(2):214-9. [ Links ]
29. Milone M, Massie R. Polymerase gamma 1 mutations: clinical correlations. Neurologist 2010;16(2):84-91. [ Links ]
30. Winterthun S, Ferrari G, He L, Taylor RW, Zeviani M, Turnbull DM, Engelsen BA, Moen G, Bindoff LA. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerasegamm mutations. Neurology 2005;64:1204-8. [ Links ]
31. Hirano M, Marti R, Spinazzola A, Nishino I, Nishigaki Y. Thymidine phosphorylase deficiency causes MNGI E: an autosomal recessive mitochondrial disorder. Nucleosides Nucleotides Nucleic Acids 2004;23(8-9):1217-25. [ Links ]
32. Tang S, Dimberg EL, Milone M, Wong L-J C. Mitochondrial neurogastrointestinal encephalomyopathy (MNGI E)-like phenotype: an expanded clinical spectrum of POLG 1 mutations. J Neurol 2012;259(5):862-8. [ Links ]
33. Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGI E, a human mitochondrial disorder. Science 1999;283(5402):689-92. [ Links ]
34. Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes - many genes, common mechanisms. Neuromuscul Disord 2010;20(7):429-37. [ Links ]
35. Blakely EL, Butterworth A, Hadden RD, Bodi I, He L, McFarland R, Taylor RW. MP V17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul Disord 2012;22(7):587-91. [ Links ]
36. Ronchi D, Garone C, Bordoni A, Gutierrez Rios P, Calvo SE, Ripolone M, Ranieri M, Rizzuti M, Villa L, Magri F, Corti S, Bresolin N, Mootha VK, Moggio M, DiMauro S, Comi GP , Sciacco M. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 2012;135(Pt 11):3404-15. [ Links ]
37. Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic communication. Biosci Rep 2007;27(1-3):39-51. [ Links ]
38. Rahman S, Poulton J. Diagnosis of mitochondrial DNA depletion syndromes. Arch Dis Child 2009;94(1):3-5. [ Links ]
39. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 2001;29(3):337-41. [ Links ]
40. Wong LJ, Brunetti-Pierri N, Zhang Q, Yazigi N, Bove KE, Dahms BB, Puchowicz, MA, Gonzalez-Gomez I, Schmitt ES, Truong CK, Hoppel CL , Chou PC , Wang J, Baldwin EE, Adams D, Leslie N, Boles RG , Kerr D, Craigen WJ. Mutations in the MP V17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology 2007;46(4):1218-27. [ Links ]
41. Nogueira C, de Souza CF, Husny A, Derks TG , Santorelli FM, Vilarinho L. MP V17: fatal hepatocerebral presentation in a Brazilian infant. Mol Genet Metab 2012;107(4):764. [ Links ]
42. El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013;10(2):186-98. [ Links ]
43. Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, DAdamo P, Calvo S, Marsano RM , Donnini C, Weiher H, Strisciuglio P, Parini R, Sarzi E, Chan A, DiMauro S, Rötig A, Gasparini P, Ferrero I, Mootha VK, Tiranti V, Zeviani M. MP V17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet 2006;38(5):570-5. [ Links ]
44. Moraes CT , Shanske S, Tritschler HJ, Aprille JR, Andreetta F, Bonilla E, Schon EA, DiMauro S. MtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet 1991;48(3):492-501. [ Links ]
45. Oskoui M, Davidzon G, Pascual J, Erazo R, Gurgel-Giannetti J, Krishna S, Bonilla E, De Vivo DC, Shanske S, DiMauro S. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol 2006;63(8):1122-6. [ Links ]
46. Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 2001;29(3):342-4. [ Links ]
47. Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet 2005;76(6):1081-6. [ Links ]
48. Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rötig A. Mutation of RRM 2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet 2007;39(6):776-80. [ Links ]
49. Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, Wibrand F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet 2007;81(2):383-7. [ Links ]
50. Nogueira C, Meschini MC , Nesti C, Garcia P, Diogo L, Valongo C, Carrozzo R, Costa R, Vilarinho L, Santorelli FM. A novel SUCL A2 mutation in a Portuguese child associated with mild methylmalonic aciduria. J Child Neurol 2013 [submitted]. [ Links ]
51. Morten KJ, Ashley N, Wijburg F, Hadzic N, Parr J, Jayawant S, Adams S, Bindoff L, Bakker HD, Mieli-Vergani G, Zeviani M, Poulton J. Liver mtDNA content increases during development: a comparison of methods and the importance of age- and tissue-specific controls for the diagnosis of mtDNA depletion. Mitochondrion 2007;7(6):386-95. [ Links ]
52. DiMauro S. Mitochondrial diseases. Biochim Biophys Acta 2004;1658(1-2):80-8. [ Links ]
53. Vasta V, Ng SB, Turner EH, Shendure J, Hahn SH . Next generation sequence analysis for mitochondrial disorders. Genome Med 2009;1(10):100. [ Links ]
54. Calvo SE, Compton AG, Hershman SG , Lim SC , Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, Christodoulou J, Fletcher JM, Bruno DL, Goldblatt J, Dimauro S, Thorburn DR, Mootha, VK. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012;4(118):118ra10. [ Links ]
55. Haack TB, Haberberger B, Frisch EM, Wieland T, Luso A, Gorza M, Strecker V, Graf E, Mayr JA, Herberg U, Hennermann JB, Klopstock T, Kuhn KA, Ahting U, Sperl W, Wilichowski E, Hoffmann GF, Tesarova M, Hansikova H, Zeman J, Plecko B, Zeviani M, Wittig I, Strom TM , Schuelke M, Freisinger P, Meitinger T, Prokisch H. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet 2012;49(4):277-83. [ Links ]
Recursos web
Human gene mutation database: HGM D Professional database: www.hgmd.cf.ac.uk/
POLG mutation database: http://tools.niehs.nih.gov/polg/index.cfm?do=polg.home&CFID=93263961&CFTOKEN=71342957
Laura Vilarinho
Instituto Nacional de Saúde Doutor Ricardo Jorge, Departamento de Genética Humana – Unidade de Investigação & desenvolvimento. Rua Alexandre Herculano, 321, 4000-055 Porto. E-mail: laura.vilarinho@insa.min-saude.pt
Data de recepção / reception date: 10-10-2013
Data de aprovação / approval date: 07-01-2014

