










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Imunoalergologia
versão impressa ISSN 0871-9721
Rev Port Imunoalergologia vol.20 no.1 Lisboa jan. 2012
Stress e doença alérgica: Mecanismos subjacentes
Gisela Calado, Graça Loureiro
Serviço de Imunoalergologia, Hospitais da Universidade de Coimbra
RESUMO
O stress é reconhecido como um factor importante de exacerbação, agravamento e diminuição da resposta ao tratamento de várias doenças alérgicas, nomeadamente da conjuntivite, rinite, asma e dermatite atópica. Contudo, os seus mecanismos etiopatogénicos continuam mal esclarecidos. Neste artigo revêem-se os mecanismos potencialmente responsáveis pelo impacto do stress sobre a doença alérgica, abordando-se o papel do sistema neuroendócrino e das suas interacções com o sistema imune, a interferência com a via oxidativa, a influência da corticorresistência e a importância dos factores genéticos.
Palavras-chave: Corticorresistência, doença alérgica, genética, sistema imune, sistema neuroendócrino, stress, via oxidativa.
Stress and allergic disease: Underlying mechanisms
ABSTRACT
Stress is recognized as an important factor of exacerbation, worsening and lack of treatment response in several allergic diseases, namely in conjunctivitis, rhinitis, asthma and atopic dermatitis. However, its etiopathogenic mechanisms are still poorly understood. In this article, the potential mechanisms responsible by the impact of stress in allergic disease are reviewed, addressing the role of the neuroendocrine system and its interactions with the immune system, the interference with the oxidative pathway, the impact of corticoresistance and the importance of genetic factors.
Key-words: Allergic disease, corticoresistance, genetics, immune system, neuroendocrine system, oxidative pathway, stress.
INTRODUÇÃO
O termo stress refere-se à resposta adaptativa do organismo perante um estímulo gerador de tensão ou sobrecarga, perigo de descompensação ou perda da homeostase. Deste modo, o stress pode ser físico, psicológico ou uma combinação dos dois. Dependendo da sua duração, pode definir-se como agudo (horas ou dias) ou crónico (meses a anos).
O stress foi inicialmente considerado por Hans Selye1, em 1936, um denominador comum a todas as reacções adaptativas do organismo, não sendo directamente lesivo para a saúde. Só mais tarde surgiria o conceito de stress como indutor de alterações fisiopatológicas (stress distress), definindo-se esta resposta como uma reacção psicofisiológica a estímulos nocivos físicos ou psicológicos1,2.
Antes da sua base inflamatória ter sido descoberta na segunda metade do século XX, as doenças alérgicas eram consideradas psicogénicas, tendo a asma e a dermatite atópica sido incluídas entre as sete doenças psicossomáticas clássicas3. Mais tarde, George Salomón foi o primeiro a afirmar a interferência dos estados emocionais (stress psicológico) com as células NK, aumentando a susceptibilidade a infecções; posteriormente, demonstrou correlação entre o nível de stress e a resposta linfoproliferativa4.
O reconhecimento da interregulação recíproca entre os sistemas nervoso e imunológico deu origem a um novo campo de estudo, a Psiconeuroimunologia. De facto, o stress crónico tem sido associado a múltiplas patologias, nomeadamente cardiovasculares (hipertensão arterial, arteriosclerose, miocardiopatia isquémica), psiquiátricas (depressão), endócrinas (diabetes mellitus, dislipidemia), gastrintestinais (síndrome do cólon irritável), neurológicas (acidentes cerebrovasculares) e infecciosas5. O inverso também ocorre, com as doenças orgânicas a influenciarem o humor e a desencadearem stress, verificando-se assim uma relação bidireccional6.
A resposta ao stress depende de vários factores, nomeadamente do tempo (stress agudo versus crónico) e intensidade de exposição, do tipo de stress mas também da idade, do sexo, da personalidade, da existência de psicopatologia prévia, dos mecanismos de defesa cognitivos e comportamentais, da predisposição genética, entre outros.
O stress é reconhecido como um factor importante no desencadeamento, agravamento e diminuição da resposta ao tratamento de várias doenças alérgicas, nomeadamente da conjuntivite, rinite, asma e dermatite atópica.
Contudo, os seus mecanismos etiopatogénicos continuam mal esclarecidos. Neste artigo os autores pretendem rever os conhecimentos actuais relativos aos mecanismos potencialmente envolvidos no impacto do stress sobre a doença alérgica.
MECANISMOS
O stress induz uma série de respostas fisiológicas, emocionais e comportamentais com o intuito de proteger o hospedeiro e de restabelecer a homeostase. Quando as respostas são excessivas ou prolongadas, como acontece na exposição crónica ao stress, podem tornar-se deletérias para o organismo7,8. Os mecanismos que, de forma independente ou interligada, ajudam a explicar a relação entre o stress e a doença alérgica, têm sido alvo de grande interesse.
Stress e sistema neuroendócrino
A intercomunicação entre o sistema nervoso central (SNC) e o sistema imune é feita através do eixo hipotálamo-hipófise-suprarrenal (HHSR) e do sistema nervoso autónomo (SNA). Esta comunicação é mediada pela acção de hormonas (adrenalina e corticosteróides), neurotransmissores (acetilcolina, noradrenalina (NA), serotonina, histamina, ácido glutâmico, ácido gama-aminobutírico (GABA), neuropéptidos (hormona adrenocorticotrófica (ACTH), hormona antidiurética (ADH), prolactina, bradicinina, somatostatina, péptido intestinal vasoactivo (VIP), substância P (SP), neuropéptido Y, encefalina, endorfina), neurotrofinas (factor de crescimento do nervo – NGF) e citocinas9(Figura 1).
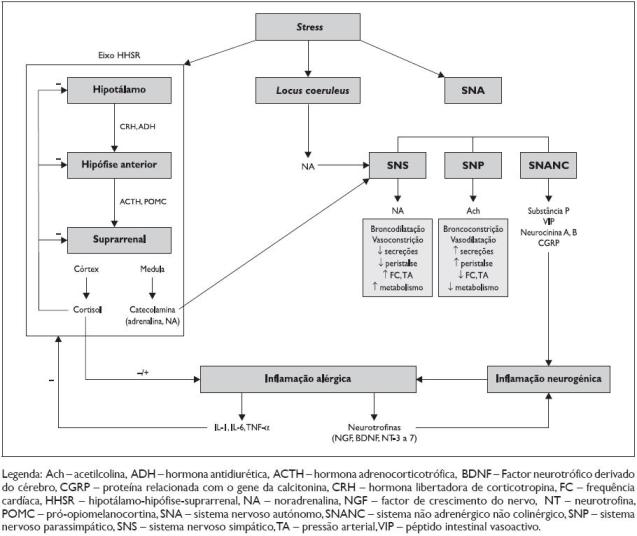
Figura 1. Stress e sistema neuroendócrino
A percepção de stress induz a activação do núcleo paraventricular do hipotálamo, com secreção de hormona libertadora de corticotropina (CRH) e de ADH, bem como a activação do locus coeruleus, centro produtor de NA e que também é activado pela CRH5. A CRH e a ADH activam o eixo HHSR, estimulando a produção de ACTH pelo lobo anterior da hipófise, com consequente activação da secreção de cortisol e catecolaminas (adrenalina e NA) pelo córtex e medula da glândula suprarrenal, respectivamente5.
Este aumento de cortisol e catecolaminas permite uma rápida mobilização de energia e oxigénio para os locais necessários, nomeadamente para os músculos e cérebro.
O cortisol, com a sua acção anti-inflamatória, tem ainda um papel fundamental na defesa contra os efeitos deletérios da resposta pró-inflamatória que é activada pelo stress10.
Em resposta ao stress prolongado, a expressão de RNAm da CRH no hipotálamo e da pró–opiomelanocortina (POMC) na hipófise anterior, precursor da ACTH, aumentam simultaneamente a hipersecreção de CRH e ACTH. Contudo, a exposição continuada à CRH induz uma diminuição da expressão dos receptores CRH-R1 na hipófise por feedback negativo, com consequente diminuição da produção de ACTH em resposta à CRH e diminuição da secreção de cortisol7,11. Apesar de a expressão basal de ADH pelo hipotálamo ser baixa, o seu aumento em resposta ao stress crónico é substancial. Deste modo, na presença de CRH (mas não de forma isolada), a ADH actua sinergicamente para potenciar a produção de ACTH através da ligação a um receptor da ADH, sem afectar a transcrição de POMC, permitindo manter a resposta da ACTH a novos estímulos indutores de stress, apesar da diminuição dos receptores CRH-R1 secundária à activação prolongada do eixo HHSR11,12.
Na exposição aguda ao stress, o aumento do cortisol contraria o agravamento da inflamação alérgica7. Alguns autores têm reportado um menor aumento dos níveis de cortisol em resposta ao stress em doentes alérgicos, comparativamente a controlos saudáveis13, embora não haja estudos prospectivos que esclareçam se esta diferença é causa ou consequência da doença alérgica. Outros autores mostraram que crianças e adultos com resolução mais rápida das exacerbações de asma alérgica apresentam uma maior elevação dos níveis de cortisol durante a exacerbação, comparativamente àqueles com resolução mais lenta14,15.
Estes estudos deixam patente a importância da integridade do eixo HHSR no curso da doença alérgica, podendo justificar um maior impacto negativo do stress nos doentes com supressão deste eixo. O cortisol tem ainda um papel regulador fundamental sobre o eixo HHSR: diminui a síntese e secreção de CRH e ACTH por feedback negativo sobre o hipotálamo e a hipófise, respectivamente16; diminui a expressão de CRH-R1 na hipófise, ocorrendo simultaneamente a diminuição da resposta destes ao efeito estimulador da CRH16; ao ligar-se aos receptores de corticosteróides (GR) presentes no hipocampo, regula negativamente o núcleo paraventricular do hipotálamo e o eixo HHSR17.
A activação do SNA é outro pilar fundamental na resposta ao stress e na intercomunicação entre o SNC e o sistema imune, uma vez que inerva órgãos e sistemas com uma importante função no sistema imune, nomeadamente fígado, baço, timo, medula óssea, gânglios linfáticos, pele, sistemas digestivo e respiratório. O SNA é constituído pelo sistema nervoso simpático (SNS) ou noradrenérgico, pelo sistema nervoso parassimpático (SNP) ou colinérgico, e pelo sistema não adrenérgico não colinérgico (SNANC) ou peptidérgico. A NA e a acetilcolina são os neurotransmissores do SNS e SNP, respectivamente, pelo que a NA secretada pelo locus coeruleus contribui para a activação do SNS. O SNS e o SNP têm acções antagónicas e é do equilíbrio entre estes dois sistemas que resulta o funcionamento normal dos órgãos. Enquanto o SNS é responsável pela mobilização de energia e oxigénio necessários à resposta de luta ou fuga perante uma situação de stress (aumenta a frequência cardíaca, a pressão arterial e o metabolismo, diminui as secreções e a peristalse, induz vasoconstrição e broncodilatação), o SNP tem os efeitos opostos, induzindo aumento das secreções, vasodilatação e edema e sendo o principal responsável pela broncoconstrição18. O SNANC tem vários mediadores, nomeadamente o VIP, a SP, as neurocininas A e B e o péptido relacionado com o gene da calcitonina (CGRP), particularmente envolvidos na inflamação neurogénica (como abordado em secção própria).
Stress e sistema imune
A activação dos sistemas nervoso e endócrino pelo stress influencia fortemente o comportamento das células do sistema imune. A CRH, produzida pelo hipotálamo e perifericamente pelos tecidos lesados, pode ligar-se aos receptores de CRH presentes na superfície mastocitária e induzir a sua desgranulação, com a libertação de citocinas e mediadores pró-inflamatórios19. Por outro lado, os próprios mastócitos sintetizam e secretam CRH em resposta à estimulação do seu receptor FcεRI20.
Alguns estudos sugerem que a desregulação do equilíbrio Th1/Th2 no sentido Th2 pode ser um mecanismo importante nas alterações imunes induzidas pelo stress crónico21. Os corticosteróides e as catecolaminas, ao inibirem a produção de IL-12 pelas células apresentadoras de antigénios (o principal indutor da resposta Th1 por estimular a secreção de IFN-γ pelas células T e NK), estimulam o predomínio do perfil Th2, com produção de IL-4, IL-10 e IL-1322,23. O VIP, conhecido como neuropéptido anti-inflamatório, também estimula o perfil Th2. Além do mecanismo referido, o VIP induz a expressão de moléculas coestimuladoras em macrófagos inactivados e em células dendríticas imaturas, além de poder exercer a sua acção directamente sobre os linfócitos T CD4+, promovendo a diferenciação Th2 24. Também as neurotrofinas contribuem para o desvio do equilíbrio Th1/Th2 no sentido Th2, podendo contribuir para o agravamento da inflamação alérgica em resposta ao stress, bem como a reactivação de vírus latentes em doentes sob maior stress psicológico, o que já foi demonstrado in vitro25.
As neurotrofinas e os neuropéptidos são os principais mediadores da inflamação neurogénica (Quadro 1), uma resposta inflamatória resultante da acção moduladora destes neuromediadores sobre as células imunes e/ou estruturais26. Da estimulação das terminações nervosas sensoriais em resposta ao stress e a estímulos químicos (produzidos durante a inflamação e lesão tecidual) resulta a secreção de neuropéptidos pró-inflamatórios que medeiam a função excitatória do SNANC por ligação a três receptores específicos das taquicininas: NK-1 (específico para a SP), NK-2 (específico para a neurocinina A) e NK-3 (específico para a neurocinina B)27. O VIP medeia a função inibitória do SNANC24. Os neuropéptidos são degradados rapidamente pela acção de peptidases, nomeadamente a endopeptidase neutra (presente nas mucosa e submucosa das vias aéreas) e a enzima conversora da angiotensina (presente nas células vasculares)28. A degradação rápida dos neuropéptidos obriga a que as suas acções sejam limitadas no tempo e restritas ao local onde são produzidas29, sendo considerados importantes na iniciação da inflamação neurogénica30,31. Contudo, a diminuição da actividade das peptidases contribui para a perpetuação deste processo inflamatório. Curiosamente, apesar de em resposta ao stress crónico as neurotrofinas, que actuam como factores de crescimento do nervo32, diminuirem em várias áreas cerebrais de ratos33, o mesmo não se verifica nos locais de inflamação alérgica, onde a sua expressão aumenta34. Num modelo murino de asma, Hahn t al demonstraram que a IL-1β, o TNF-α e as citocinas Th2estimulam fortemente a produção do factor de crescimento do nervo (NGF) e do factor neurotrófico derivado do cérebro (BDNF) pelas células epiteliais das vias aéreas, o que não acontece com o IFN-γ, uma citocina Th135. Apesar de a exposição crónica ao stress não ter um efeito estimulador directo sobre as neurotrofinas, a inflamação neurogénica (mediada pelos neuropéptidos secretados em resposta ao stress) poderá ser responsável por induzir a produção local de neurotrofinas. Assim, apesar de em condições fisiológicas as células do sistema nervoso serem a fonte e alvo primários dos neuropéptidos e neurotrofinas, durante a inflamação alérgica as células estruturais (queratinócitos, células epiteliais, endoteliais e do músculo liso, células das glândulas submucosas) e do sistema imune (monócitos, macrófagos, mastócitos, linfócitos T, eosinófilos) também os podem produzir, constituindo também o seu alvo ao expressarem os seus receptores36-40. As neurotrofinas actuam por ligação aos seguintes receptores de superfície celular: p75NTR (receptor pan-neurotrofina ao qual se ligam, com baixa afinidade e especificidade, todas a neurotrofinas maduras), TrkA (tropomyosine-related kinase A, reconhecido especificamente pelo NGF), TrkB (activado pelo BDNF, neurotrofina 4 e 5) e TrkC (reconhecido primariamente pela neurotrofina 3)41.
Quadro 1. Principais mediadores da inflamação neurogénica

A produção local intensa e contínua de neurotrofinas durante a inflamação alérgica (por células estruturais e imunes) estimula a secreção de neuropéptidos pelas terminações nervosas sensoriais42, influenciando a intensidade e duração das respostas imunes locais. Deste modo, as neurotrofinas são importantes na amplificação e manutenção da inflamação neurogénica, um ciclo vicioso de interacções neuroimunes que potenciam a inflamação alérgica26,35,42. A inflamação neurogénica resulta do efeito vasodilatador importante dos seus mediadores (a SP é um dos vasodilatadores conhecidos mais potente, 100 vezes superior à histamina em concentrações molares idênticas43) e da acção moduladora dos neuropéptidos e das neurotrofinas sobre as células do sistema imune, as quais têm sido amplamente referidas na literatura40. A SP actua sobre monócitos e macrófagos aumentando a produção de TNF-α e IL-12. Tem ainda a capacidade de induzir directamente a desgranulação dos mastócitos44. O NGF tem uma importante acção moduladora sobre mastócitos, recrutando-os, influenciando o seu desenvolvimento e promovendo a sua sobrevivência como cofactor do stem cell factor45,46. Num modelo murino de doença respiratória alérgica, o bloqueio da sinalização do NGF pela aplicação intranasal de anticorpos neutralizantes inibiu a resposta alérgica inicial induzida pelo alergénio, a qual é mediada pela desgranulação dos mastócitos 47. Devido à sua acção antiapoptótica, as neurotrofinas promovem a sobrevivência dos eosinófilos durante a inflamação alérgica34,35,48, contribuindo para a eosinofilia, uma fonte importante de citocinas inflamatórias (ex. IL-4, IL-13) e quimiocinas (ex. RANTES, MCP-1), além de estimular a produção de mucina35. Alguns estudos demonstraram a actividade quimiotática sobre eosinófilos do NGF, BDNF, neurotrofina-3 e neurotrofina-435,49. De um modo geral, a inflamação neurogénica contribui para as manifestações de doença alérgica em diferentes órgãos (olhos, vias aéreas, pele) resultando em vasodilatação, aumento da permeabilidade vascular e edema, prurido, recrutamento e activação de células imunes. Nas vias aéreas aumenta a secreção de muco e ainda induz broncoconstrição por aumento da actividade colinérgica18, a qual é estimulada por taquicininas50. As neurotrofinas também podem induzir hiperreactividade brônquica (HRB), na presença e na ausência de inflamação das vias aéreas, como demonstrado num modelo murino tratado com NGF em que a HRB foi induzida sem inflamação prévia das vias aéreas51. O NGF também parece estar envolvido na remodelação das vias aéreas ao induzir a produção de colagénio e migração de fibroblastos e a sua diferenciação em miofibroblastos52,53; tem ainda um importante papel no aumento da vascularização ao induzir a proliferação de células endoteliais e células do músculo liso vascular e ao estimular a libertação de factores pró-angiogénicos54. Um modelo murino de asma sugeriu o papel directo das neurotrofinas na indução de um perfil Th2, uma vez que o tratamento com anticorpos anti-NGF diminuiu a função das células Th2 e que o receptor TrkC foi encontrado nestas células mas não em Th1 55. Também o BDNF demonstrou aumentar a produção de IgE específica e reduzir as citocinas de tipo Th1, favorecendo a resposta Th2 56.
Estudos recentes in vitro sugerem que a SP pode induzir um fenótipo Th17 em indivíduos com distúrbio de ansiedade57. O mecanismo implicado não é conhecido mas os mesmos autores especulam que a regulação insuficiente por células T reguladoras (Treg) possa estar envolvida, uma vez que os níveis de IL-10 e de IL-2 (citocina essencial na homeostase de Treg in vivo 58 e na sua função imunossupressora n vitro59) estavam diminuidos. As células Th17 têm um papel importante na defesa contra bactérias extracelulares e fungos60, promovendo uma intensa quimiotaxia de neutrófilos para os locais de inflamação através da secreção de IL-8 por fagócitos e células locais (activadas pelas citocinas IL-17, IL-21, IL-1β,IL-6, TNF-α), o que torna o processo inflamatório resistente à acção dos corticosteróides. A interacção entre o sistema nervoso e o sistema imune é bidireccional, já que o sistema imune também modula a função do sistema nervoso. Um dos mecanismos resulta das citocinas secretadas, nomeadamente em resposta às infecções víricas. As infecções víricas podem agravar a resposta alérgica por diferentes mecanismos: são o desencadeante de exacerbação de asma mais frequente61, podem causar HRB inespecífica62, induzem a secreção de citocinas com acção sobre o eixo HHSR63,64. A presença de vírus em células infectadas é reconhecida pela detecção de moléculas de RNA de cadeia dupla sintetizadas durante a replicação viral. Este sinal leva à transcrição e secreção de interferões (IFN)-tipo I (IFN-α e IFN-β), TNF-α, IL-1 e IL-6 por células imunes activadas, nomeadamente macrófagos (e microglia no SNC), células endoteliais, fibroblastos e neurónios. Este ambiente pró-inflamatório e antiviral protege as células vizinhas ainda não infectadas, tornando-as resistentes à replicação viral. Os IFN-tipo I activam ainda as células NK (função lítica; quando estimuladas por IL-12 secretam IFN-γ (IFN tipo II), crucial no controlo da infecção antes da activação das células T) e aumentam a expressão de MHC classe I, facilitando a apresentação de antigénio às células T CD8+. Na presença de IL-12 (secretada por macrófagos e células dendríticas) e IFN-γ, as células T CD4+ diferenciam-se no sentido Th1, promovendo a imunidade celular através da activação de macrófagos (secreção IFN-γ) e células T CD8+(secreção de IL-2). Estas citocinas ligam-se a receptores presentes a vários níveis do eixo HHSR, estimulando a produção de cortisol por acção sobre o hipotálamo (mecanismo principal) ou por acção directa sobre a hipófise ou suprarrenal63. Há ainda citocinas produzidas localmente pelo cérebro, hipófise anterior e suprarrenal, cuja acção parácrina parece amplificar e manter a actividade do eixo HHSR durante a inflamação crónica63. A secreção adicional de citocinas pelas células do sistema imune é, em circunstâncias normais, inibida por feedback negativo do cortisol, protegendo o organismo de uma resposta antiviral exagerada (lesão tecidual, resposta auto-imune ou choque séptico)63.
Stress e via oxidativa
O desequilíbrio entre espécies reactivas de oxigénio (ERO) e antioxidantes denomina-se stress oxidativo. A denominação ERO resulta da sua elevada capacidade de oxidar proteínas, lípidos e ADN, levando a danos estruturais importantes. As ERO são ubiquitárias no meio intra e extracelular, sendo produzidas por fontes endógenas (ex. produtos do metabolismo celular) e exógenas (ex. poluição atmosférica, fumo de tabaco)65. O stress psicológico tem sido apontado como um factor ambiental adicional com acção pró-oxidativa66, embora a forma como isso acontece não esteja esclarecida. Os antioxidantes endógenos são a primeira linha de defesa contra as ERO, incluindo enzimas antioxidantes (ex. famílias da superóxido dismutase, catalase, glutationa-peroxidase, glutatião S-transferase e tioredoxina) e mecanismos não enzimáticos (de baixo peso molecular: glutationa, ascorbato, urato, bilirrubina, ácido lipóico; de alto peso molecular: albumina, proteínas ligadas a metais livres, como a transferrina)65. O equilíbrio dinâmico entre a produção de ERO e a resposta antioxidante é fundamental, sob pena de haver dano celular. Li N t al propuseram um modelo hierárquico para explicar a resposta ao stress oxidativo (Figura 2) 67,68. Assim, baixos níveis de stress oxidativo (Nível 1) levam à transactivação do factor de transcrição factor nuclear derivado de eritróide 2 (Nrf-2), responsável pela activação da transcrição de mais de 200 genes com funções antioxidantes, anti-inflamatórias e citoprotectoras.
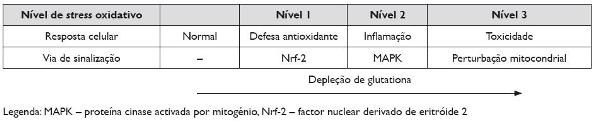
Figura 2. Modelo hierárquico da resposta ao stress oxidativo (adaptado da referência 68)
Quando a capacidade de defesa desta via é ultrapassada por uma maior produção de ERO, ocorre a activação de outras cascatas de sinalização intracelular potencialmente lesivas, como é o caso da activação da proteína cinase activada por mitogénio (MAPK) e do factor nuclear kB (NF-kB) (Nível 2), os quais induzem a expressão de agentes pró-inflamatórios, nomeadamente citocinas (IL-4, IL-5, IL-8, IL-10, IL-13, TNF-α), quimiocinas (MIP-1α, MCP-3, RANTES) e moléculas de adesão (ICAM-1, VCAM-1). Ao aumentar a activação e recrutamento de células efectoras através dos produtos libertados, esta resposta de defesa contribui para perpetuar a inflamação alérgica. Um nível ainda superior de stress oxidativo pode, em último caso, desencadear uma resposta citotóxica com origem na mitocôndria, culminando em apoptose celular ou necrose (Nível 3). O papel desta última resposta na patogénese da doença alérgica é desconhecido.
O stress oxidativo, além da sua acção pró-inflamatória, parece ter um papel adjuvante na sensibilização a aeroalergénios69,70. Este papel pode ser explicado, pelo menos em parte, pelo seu impacto sobre as células apresentadoras de antigénios, nomedamente as células dendríticas (CD). Num estudo com CD murinas, o stress oxidativo resultante da exposição a substâncias químicas eliminadas por um tubo de escape inibiram a sua maturação induzida pela ligação de lipopolissacáridos (LPS) aos Toll-like receptors (TLR), bem como a secreção de IL-1271. Este efeito pode interferir na produção de IFN-γ pelas células T, suprimindo a diferenciação Th1 e favorecendo um desvio Th2. Outro estudo em modelo animal mostrou que a diminuição dos níveis de glutationa nas CD em resposta ao stress oxidativo diminuiu a resposta Th1, mas que o tratamento destas CD com N-acetilcisteína, um precursor tiol, restaurou a imunidade Th1 72,73.
Stress e corticorresistência
Os corticosteróides, pela sua potente acção anti-inflamatória, são o grupo farmacológico mais usado no tratamento das doenças alérgicas, induzindo ou inibindo a expressão de genes-alvo. Contudo, sabe-se que um subgrupo de doentes não responde à corticoterapia apesar do uso de altas doses, o que resulta da interacção de vários factores, nomeadamente genéticos, ambientais e imunológicos74.
Admite-se que a exposição crónica ao stress, ao activar o eixo HHSR de forma prolongada, pode resultar numa resposta contra-reguladora, com subregulação da expressão e/ou funcão dos GR, levando a uma resistência funcional adquirida7,11,74,75. Num estudo in vitro, Miller et al observaram que as células mononucleares circulantes de adolescentes e pré-adolescentes asmáticos com famílias disfuncionais eram mais resistentes aos efeitos da hidrocortisona quanto à expressão de citocinas (IL-5 e IFN-γ) e à activação de eosinófilos, comparativamente àqueles com maior apoio parental76. A subregulação do gene que codifica o GR poderá, pelo menos em parte, justificar este fenómeno de corticor resistência74-76. Outro mecanismo possível é a incapacidade de o GR sofrer translocação para o núcleo ou de se ligar ao ADN, onde interfere com a expressão génica77. A diminuição do RNAm do GR e da sua expressão proteica também foram descritos78. Não está claro se a diminuição do RNAm resulta da supressão da activação do promotor, de diminuição da sua estabilidade ou de ambos79. A expressão do GR é subregulada pela ligação do cortisol em situações em que o stress e a inflamação alérgica coexistem, mas outras causas podem estar envolvidas, nomeadamente a inibição da expressão do gene da isoforma GRα pelo factor nuclear-kB (NF-kB) nos tecidos inflamados79,80. Bailey MT et al mostraram que a expressão aumentada do NF-kB no núcleo de células do sistema imune ocorre em simultâneo com a incapacidade de o GR sofrer translocação para o núcleo, de se ligar ao ADN e com a diminuição da expressão do GR78. Apesar de a expressão do GR ser ubiquitária, a regulação do gene GR induz diferenças na expressão do seu RNAm e da sua proteína em diferentes subtipos celulares81. Alguns estudos têm demonstrado que o impacto do stress crónico sobre a produção de citocinas dependente dos TLR, nomeadamente do TLR-4, também pode contribuir para esta corticorresistência82,83. Também as vias do stress oxidativo têm sido implicadas na asma corticorresistente pelo predomínio de inflamação neutrofílica (e não eosinofílica), como observado num modelo murino de exacerbação de asma84.
Noutra perspectiva, estudos de genética e epigenética têm demonstrado que a exposição a uma resposta alterada dos GR durante a fase inicial do desenvolvimento (incluindo in utero) induz grandes alterações nos mecanismos neuroendócrinos e imunes, podendo potenciar o risco de asma85.
Stress e genética
Alguns estudos demonstraram a influência dos factores genéticos nos níveis basais de cortisol e nas suas variações ao acordar em adultos e crianças, o que pode interferir na actividade do eixo HHSR86. Polimorfismos do gene do GR modificam a sensibilidade ao cortisol produzido em resposta ao stress, afectando também o eixo HHSR21,87. Num estudo com crianças asmáticas, Miller GE et al observaram que a exposição crónica ao stress diminuiu a expressão dos genes codificadores dos GR e dos receptores adrenérgicos β2 88. O mesmo não se verificou com a exposição aguda isolada, embora quando sobreposta à exposição crónica tenha acentuado o impacto sobre a expressão génica. Estas crianças sob stress agudo e crónico simultâneo apresentaram uma redução de 5,5 vezes do RNAm do GR e de 9,5 vezes do RNAm do receptor adrenérgico β2, comparativamente a crianças asmáticas sem esta exposição. Também os polimorfismos dos genes que codificam factores envolvidos nos mecanismos de feedback na resposta dos glicocorticóides ao stress, tais como o receptor da CRH (genes CRHR1 e CRHR2), o TNF-α e outros loci de citocinas, parecem estar envolvidos86,87. A variante CRHR1, o receptor predominante na hipófise e pulmões, tem sido associada a uma melhor resposta à corticoterapia inalada89.
Polimorfismos genéticos de enzimas antioxidantes podem contribuir para a ineficácia deste mecanismo de defesa (ex. polimorfismo da glutationa-S-transferase (GST)M1 e GSTP1). Num estudo com crianças asmáticas verificou-se sintomatologia respiratória mais importante nas crianças com os genótipos GSTM1 null ou GSTP1 Val/Val. Na presença dos dois genótipos, os sintomas respiratórios eram ainda mais graves90. Curiosamente, os polimorfismos referidos estão presentes em aproximadamente 50% e 40% da população, respectivamente, o que revela o seu impacto na saúde91.
Stresse doença alérgica
O stress é reconhecido como um factor com impacto negativo sobre a doença alérgica, nomeadamente na conjuntivite37,92, rinite93,94, asma7,14,15,30,31,88,95,96 e dermatite atópica24,97,98. Embora se tenha abordado cada um dos mecanismos de forma independente, a interacção entre o stress e a doença alérgica é extremamente complexa, verificando-se uma importante interligação e interregulação entre os diferentes mecanismos. A importância do sistema neuroimune-endócrino está bem definida e foi alvo de consideração neste artigo. Mais recentemente foi descrito o sistema neuroimune-cutâneo, particularmente relevante na inflamação neurogénica a nível cutâneo e cujo papel na patogénese das doenças cutâneas alérgicas e inflamatórias tem assumido um impacto crescente. Este tema foi revisto por outros autores, ultrapassando o objectivo deste artigo24,98.
CONCLUSÃO
Não há dúvida que a relação entre os sistemas nervoso e imune é real, complexa e bidirecional. A dificuldade em quantificar o impacto clínico da exposição crónica ao stress na doença alérgica leva a que na maioria das vezes este seja menosprezado. Os mecanismos que, de forma independente ou interligada, ajudam a explicar esta relação têm sido alvo de grande interesse, havendo ainda muito por descobrir. Os conhecimentos actuais sugerem que, em indivíduos geneticamente predispostos, a exposição crónica ao stress pode favorecer a expressão de doença alérgica, bem como complicar o controlo da mesma. A melhor elucidação dos mecanismos responsáveis pela interação entre o stress e a doença alérgica poderá ajudar a identificar novos alvos terapêuticos, melhorando a abordagem da doença alérgica.
REFERÊNCIAS
1 Selye H. A syndrome produced by diverse nocuous agents. Nature 1936; 138: 32. [ Links ]
2 Selye H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocrinol 1946; 6: 117-230. [ Links ]
3 Alexander F. Psychosomatic medicine – Its Principles and applications. New York: Norton; 1950. [ Links ]
4 Gómez-Magaña PA, Ongay-Pérez A. Aspectos psiquiátricos de la alergia. In: Méndez J (Ed.). Alergia: enfermedad multisistémica. Fundamentos básicos y clínicos. Ciudad de México: Editorial Médica Panamericana; 2008: 259-69. [ Links ]
5 Montoro J, Mullol J, Jáuregui I, Dávila I, Ferrer M, Bartra J, et al Stress and allergy. J Investig Allergol Clin Immunol 2009; 19 (Suppl 1): 40-7. [ Links ]
6 Chida Y, Hamer M, Steptoe A. A bidirectional relationship between psychosocial factors and atopic disorders: a systematic review and meta-analysis. Psychosomatic Medicine 2008; 70: 102-16. [ Links ]
7 Vig R, Forsythe P, Vliagoftis H. The role of stress in asthma. Insight from studies on the effect of acute and chronic stressors in models of airway inflammation. Ann N Y Acad Sci 2006; 1088: 65-77. [ Links ]
8 Okuyama K, Ohwada K, Sakurada S, Sato N, Sora I, Tamura G, et al The distinctive effects of acute and chronic psychological stress on airway inflammation in a murine model of allergic asthma. Allergol Int 2007; 56: 29-35. [ Links ]
9 Ziemssen T, Kern S. Psychoneuroimmunology – Cross-talk between the immune and nervous systems. J Neurol 2007; 254 (Suppl 2): II/8-11. [ Links ]
10 Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrinol Rev 1984; 5: 25-44. [ Links ]
11 Hauger RL, Hauger FM. Dautzenberg. Regulation of the stress response by corticotropin - releasing factor receptors. In: Conn PM, Freeman ME (Eds.). Neuroendocrinology in Physiology and Medicine. Totowa, NJ: Humana Press Inc; 2000: 261-86. [ Links ]
12 Kovacs KJ. Functional neuroanatomy of the parvocellular vasopressinergic system: transcriptional responses to stress and glucocorticoid feedback. Prog Brain Res 1998; 119: 31-43. [ Links ]
13 Wamboldt MZ, Laudenslager M, Wamboldt FS, Kelsay K, Hewitt J. Adolescents with atopic disorders have an attenuated cortisol response to laboratory stress. J Allergy Clin Immunol 2003;111: 509-14. [ Links ]
14 Nomura S, Fujitaka M, Sakura N, Ueda K. Adrenocortical function in asthmatic children: low levels of adrenocortical hormones in children with persistent attacks. Eur J Pediatr 1997; 156: 323-8. [ Links ]
15 Peebles RS Jr, Togias A, Bickel CA, Diemer FB, Hubbard WC, Schleimer RP. Endogenous glucocorticoids and antigen-induced acute and late phase pulmonary responses. Clin Exp Allergy 2000; 30: 1257-65. [ Links ]
16 Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocrinol Rev 1984; 5: 1-24. [ Links ]
17 Herman JP, Schafer MK, Young EA, Thompson R, Douglass J, Akil H, et al Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamo-pituitary-adrenocortical axis. J Neurosci 1989; 9: 3072-82. [ Links ]
18 Larsen GL, Fame TM, Renz H, Loader JE, Graves J, Hill M, et al Increased acetylcholine release in tracheas from allergen-exposed IgE immune mice. Am J Physiol 1994; 266: 263-70. [ Links ]
19 Theoharides TC, Singh LK, Boucher W, Pang X, Letourneau R, Webster E, et al Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology 1998; 139: 403-13. [ Links ]
20 Theoharides TC, Donelan JM, Papadopoulou N, Cao J, Kempuraj D, Conti P. Mast cells as targets of corticotrophin-releasing factor and related peptides. Trends Pharmacol Sci 2004; 25: 563-8. [ Links ]
21 Wright RJ. Stress and atopic disorders. J Allergy Clin Immunol 2005; 116: 1301-6. [ Links ]
22 DeKruyff RH, Fang Y, Umetsu DT. Corticosteroids enhance the capacity of macrophages to induce Th2 cytocine synthesis in CD4+lymphocytes by inhibiting IL-12 production. J Immunol 1998; 160: 2231-7. [ Links ]
23 Elenkov IJ, Chrousos GP. Stress hormones, Th1/Th2 patterns, pro/anti -inflammatory cytokines and susceptibility to disease. Trends Endocrinol Metabol 1999; 10: 359-68.
24 Kalil-Gaspar P. Neuropeptídeos na pele. An Bras Dermatol 2003; 78: 483-98. [ Links ]
25 Marshall GD, Agarwal SK, Glaser R. Enhancement of chronic HIV infection in vitro stress hormones. Neuroimmunomodulation 1999; 6: 235 (Abstract). [ Links ]
26 Mostafa GA. Neurogenic inflammation and allergy. Egypt J Pediatr Allergy Immunol 2009; 7: 45-58. [ Links ]
27 Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, Martin JD, et alTachykinins and tachykinin receptors: structure and activity relationships. Curr Med Chem 2004; 11: 2045-81. [ Links ]
28 Di Maria GU, Bellofiore S, Geppetti P. Regulation of airway neurogenic inflammation by neutral endopeptidase. Eur Respir J 1998; 12:1454-62. [ Links ]
29 Nadel JA. Neutral endopeptidase modulates neurogenic inflammation. Eur Respir J 1991; 4: 745-54. [ Links ]
30 Groneberg DA, Quarcoo D, Frossard N, Fischer A. Neurogenic mechanisms in bronchial inflammatory diseases. Allergy 2004; 59:1139-52. [ Links ]
31 Barnes PJ. Neurogenic inflammation in the airways. Respir Physiol 2001; 125: 145-54. [ Links ]
32 Ernfors P. Local and target -derived actions of neurotrophins during peripheral nervous system development. Cell Mol Life Sci 2001; 58:1036-44. [ Links ]
33 Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 1995; 15(3 Pt 1): 1768-77. [ Links ]
34 Rapp U, Deneka N, Bruder A, Kapp A, Wedi B. Differential up-regulation of neurotrophin receptors and functional activity of neurotrophins on peripheral blood eosinophils of patients with allergic rhinitis, atopic dermatitis and nonatopic subjects. Clin Exp Allergy 2008; 38: 1493-8. [ Links ]
35 Hahn C, Islamian AP, Renz H, Nockher WA. Airway epithelial cells produce neurotrophins and promote the survival of eosinophils during allergic airway inflammation. J Allergy Clin Immunol 2006; 117: 787-94. [ Links ]
36 Nockher WA, Renz H. Neurotrophins in inflammatory lung diseases: modulators of cell differentiation and neuroimmune interactions. Cytokine Growth Factor Rev 2003; 14: 559-78. [ Links ]
37 Micera A, Lambiase A, Bonini S. The role of neuromediators in ocular allergy. Curr Opin Allergy Clin Immunol 2008; 8: 466-71. [ Links ]
38 Bonini S, Rasi G, Bracci-Laudiero ML, Procoli A, Aloe L. Nerve growth factor: neurotrophin or cytokine? Int Arch Allergy Immunol 2003; 131: 80-4. [ Links ]
39 Joachim RA, Sagach V, Quarcoo D, Dinh QT, Arck PC, Klapp BF. Neurokinin-1 receptor mediates stress-exacerbated allergic airway inflammation and airway hyperresponsiveness in mice. Psychosom Med 2004; 66: 564-71. [ Links ]
40 Nockher WA, Renz H. Neurotrophins in allergic diseases: from neuronal growth factors to intercellular signalling molecules. J Allergy Clin Immunol 2006; 117: 583-9. [ Links ]
41 Miller FD, Kaplan DR. Neurobiology: TRK makes the retrograde. Science 2002; 295: 1471-3. [ Links ]
42 Renz H, Kerzel S, Nockhe WA. The role of neurotrophins in bronchial asthma: contribution of the pan-neurotrophin receptor p751. Prog Brain Res 2004; 146: 325-33. [ Links ]
43 Foreman JC. Peptides and neurogenic inflammation. Br Med Bulletin 1987; 43: 386-400. [ Links ]
44 OConnor TM, OConnell J, OBrien DI, Goode T, Bredin CP, Shanahan F. The role of substance P in inflammatory disease. J Cell Phy siol 2004; 201: 167-80. [ Links ]
45 Sawada J, Itakura A, Tanaka A, Furusaka T, Matsuda H. Nerve growth factor functions as a chemoattractant for mast cells through both mitogen-activated kinase and phosphatidylinositol 3 - kinase signaling pathways. Blood 2000; 95: 2052-8. [ Links ]
46 Kanbe N, Kurosawa M, Miyachi Y, Kanbe M, Saitoh H, Matsuda H. Nerve growth factor prevents apoptosis of cord blood–derived human cultured mast cells synergistically with stem cell factor. Clin Exp Allergy 2000; 30: 1113-20. [ Links ]
47 Braun A, Appel E, Baruch R, Herz U, Botchkarev V, Paus R, et al Role of nerve growth factor in a mouse model of allergic airway inflammation and asthma. Eur J Immunol 1998; 28: 3240-51. [ Links ]
48 Nassenstein C, Braun A, Erpenbeck VJ, Lommatzsch M, Schmidt S, Krug N, et al The neurotrophins nerve growth factor, brain–derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 are survival and activation factors for eosinophils in patients with allergic bronchial asthma. J Exp Med 2003; 198: 455-67. [ Links ]
49 Nockher WA, Renz H. Neurotrophins and asthma: Novel insight into neuroimmune interaction. J Allergy Clin Immunol 2006; 117: 67-71. [ Links ]
50 Fryer AD, Jacoby DB. Plasticity of cholinergic and tachykinergic nerves: the convergence of the twain. Am J Physiol Lung Cell Mol Physiol 2002 L907-8. [ Links ]
51 Braun A, Quarcoo D, Schulte-Herbrüggen O, Lommatzsch M, Hoyle G, Renz H. Nerve growth factor induces airway hyperresponsiveness in mice. Int Arch Allergy Immunol 2001; 124: 205-7. [ Links ]
52 Micera A, Vigneti E, Pickholtz D, Reich R, Pappo O, Bonini S, et alNerve growth factor displays stimulatory effects on human skin and lung fibroblasts, demonstrating a direct role for this factor in tissue repair. Proc Natl Acad Sci USA 2001; 98: 6162-7. [ Links ]
53 Nithya M, Suguna L, Rose C. The effect of nerve growth factor on the early responses during the process of wound healing. Biochim Biophys Acta 2003; 1620: 25-31. [ Links ]
54 Cantarella G, Lempereur L, Presta M, Ribatti D, Lombardo G, Lazarovici P, et al. Nerve growth factor - endothelial cell interaction leads to angiogenesis in vitro and in vivo. FASEB J 2002; 16: 1307-9. [ Links ]
55 Sekimoto M, Tsuji T, Matsuzaki J, Chamoto K, Koda T, Nemoto K, et al Functional expression of the TrkC gene, encoding a high affinity receptor for NT-3, in antigen-specific T helper type 2 (Th2) cells. Immunol Lett 2003; 88: 221-6. [ Links ]
56 Kimata H. Brain-derived neurotrophic factor selectively enhances allergen-specific IgE production. Neuropeptides 2005; 39: 379-83. [ Links ]
57 Barros PO, Ferreira TB, Vieira MM, Almeida CR, Araújo-Lima CF, Silva-Filho RG, et al. Substance P enhances Th17 phenotype in individuals with generalized anxiety disorder: an event resistant to glucocorticoid inhibition. J Clin Immunol 2011; 31: 51-9. [ Links ]
58 Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol Lett 2007; 114: 1-8. [ Links ]
59 Horwitz DA, Zheng SG, Wang J, Gray JD. Critical role of IL-2 and TGF -βin generation, function and stabilization of Foxp3+CD4+Treg. Eur J Immunol 2008; 38: 912-5. [ Links ]
60 Matsuzaki G, Umemura M. IL-17 as an effector molecule of innate and acquired immunity against infections. Microbiol Immunol 2007; 51: 1139-47. [ Links ]
61 Papadopoulos NG, Xepapadaki P, Mallia P, Brusselle G, Watelet J-B, Xatzipsalti M, al Mechanisms of virus-induced asthma exacerbations: state-of-the-art. A GA2LEN and InterAirways document. Allergy 2007;62: 457-70.
62 Everard ML. The relationship between respiratory syncycial virus infections and the development of wheezing and asthma in children. Curr Opin Allergy Clin Immunol 2006; 6: 56-61. [ Links ]
63 Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol 2005; 18: 41-78. [ Links ]
64 Lindemans CA, Kimpen JL. The immune response to viral lower respiratory tract infection. Adv Exp Med Biol 2005; 568: 41-65. [ Links ]
65 Bowler RP, Crapo JD. Oxidative stress in allergic respiratory diseases. J Allergy Clin Immunol 2002; 110: 349-56. [ Links ]
66 66. Moller P, Wallin H, Knudsen LE. Oxidative stress associated with exercise, psychological stress and life-style factors. Chem Biol Interact 1996; 102: 17-36. [ Links ]
67 Li N, Hao M, Phalen RF, Hinds WC, Nel AE. Particulate air pollutants and asthma: a paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin Immunol 2003; 109: 250-65. [ Links ]
68 Xiao GG, Wang M, Macatangay G, Li N, Loo JA, Nel AE. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particles in a macrophage cell line. J Biol Chem 2003; 278: 50781-90. [ Links ]
69 Diaz-Sanchez D, Garcia MP, Wang M, Jyrala M, Saxon A. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J Allergy Clin Immunol 1999; 104: 1183-8. [ Links ]
70 Fujieda S, Diaz-Sanchez D, Saxon A. Combined nasal challenge with diesel exhaust particles and allergen induces in vivo IgE isotype switching. Am J Respir Cell Mol Biol 1998; 19: 507-12. [ Links ]
71 Chan RC, Wang M, Li N, Yanagawa Y, Onoé K, Lee JJ, et al. Pro-oxidative diesel exhaust particle chemicals inhibit LPS – induced dendritic cell responses involved in T-helper differentiation. J Allergy Clin Immunol 2006; 118: 455-65. [ Links ]
72 Kim HJ, Barajas B, Chan RC, Nel AE. Glutathione depletion inhibits dendritic cell maturation and delayed – type hypersensitivity: implications for systemic disease and immunosenescence. J Allergy Clin Immunol 2007; 119: 1224-33. [ Links ]
73 Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci USA 1998; 95: 3071-6. [ Links ]
74 Leung DYM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol 2003; 111: 3-22. [ Links ]
75 Corrigan CJ, Loke TK. Clinical and molecular aspects of glucocorticoid resistant asthma.Therapeutics Clin Risk Manage 2007; 3: 771-87. [ Links ]
76 Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 2002; 21: 531-41. [ Links ]
77 Quan N, Avitsur R, Stark JL, He L, Lai W, Dhabhar F, et al Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol 2003; 137: 51-8. [ Links ]
78 Bailey MT, Kierstein S, Sharma S, Spaits M, Kinsey SG, Tliba O, et al Social stress enhances allergen-induced airway inflammation in mice and inhibits corticosteroid responsiveness of cytokine production. J Immunol 2009; 182: 7888-96. [ Links ]
79 Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol 2002; 83: 37-48. [ Links ]
80 Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest 2008; 134: 394-401. [ Links ]
81 Kalinyak JE, Dorin RI, Hoffman AR, Perlman AJ. Tissue-specific regulation of glucocorticoid receptor mRNA by dexamethasone. J Biol Chem 1987; 262: 10441-4. [ Links ]
82 Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, et al Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun 2009; 23: 225-31. [ Links ]
83 Zhang Y, Zhang Y, Miao J, Hanley G, Stuart C, Sun X, et al Chronic restraint stress promotes immune suppression through toll – like receptor 4 -mediated phosphoinositide 2 -kinase signalling. J Neuroimmunol 2008; 204: 13-9. [ Links ]
84 Ito K, Herbert C, Siegle JS, Vuppusetty C, Hansbro N, Thomas PS, et al Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am J Respir Cell Mol Biol 2008; 39: 543-50. [ Links ]
85 Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008; 3: 97-106. [ Links ]
86 Wust S, Federenko IS, van Rossum EF, Koper JW, Kumsta R, Entringer S, et al A psychobiological perspective on genetic determinants of hypothalamus-pituitary -adrenal axis activity. Ann N Y Acad Sci 2004; 1032: 52-62. [ Links ]
87 Wust S, Van Rossum EF, Federenko IS, Koper JW, Kumsta R, Hellhammer DH. Common polymorphisms in the glucocorticoid receptor gene are associated with adrenocortical responses to psychosocial stress. J Clin Endocrinol Metab 2004; 89: 565-73. [ Links ]
88 Miller GE, Chen E. Life stress and diminished expression of genes encoding glucocorticoid receptor and β 2-adrenergic receptor in children with asthma. Proc Natl Acad Sci USA 2006; 103: 5496-501. [ Links ]
89 Tantisira KG, Lake S, Silverman ES, Palmer LJ, Lazarus R, Silverman EK, et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatic treated with inhaled corticosteroids. Hum Mol Genet 2004; 13: 1353-9. [ Links ]
90 Romieu I, Ramirez-Aguillar M, Sienra-Monge JJ, Moreno-Macías H, del Rio -Navarro BE, David G, et al. GSTM1 and GSTP1 and respiratory health in asthmatic children exposed to ozone. Eur Respir J 2006; 28: 953-9. [ Links ]
91 Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL, et alMetabolic gene polymosphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev 2001; 10: 1239-48. [ Links ]
92 Amod R. Immunology of allergic eye disease. Curr Allergy Clin Immunol 2006;9:70-3. [ Links ]
93 Pfaar O, Raap U, Holz M, Hörmann K, Klimek L. Pathophysiology of itching and sneezing in allergic rhinitis. Swiss Med Wkly 2009; 139: 35-40. [ Links ]
94 Wu X, Myers AC, Goldstone AC, Togias A, Sanico AM. Localization of nerve growth factor and its receptors in the human nasal mucosa. J Allergy Clin Immunol 2006; 118: 428-33. [ Links ]
95 Nassenstein C, Schulte-Herbruggen O, Renz H, Braun A. Nerve growth factor: the central hub in the development of allergic asthma? Eur J Pharmacol 2006; 533: 195-206. [ Links ]
96 Butler CA, Heaney LG. Neurogenic inflammation and asthma. Inflamm Allergy Drug Targets 2007; 6: 127-32. [ Links ]
97 Arndt J, Smith N, Tausk F. Stress and atopic dermatitis. Allergy Asthma Rep 2008; 8: 312-17. [ Links ]
98 Cevikbas F, Steinhoff A, Homey B, Steinhoff M. Neuroimmune interactions in allergic skin diseases. Curr Opin Allergy Clin Immunol 2007; 7: 365-73. [ Links ]
Gisela Calado
Serviço de Imunoalergologia
Hospitais da Universidade de Coimbra
Praceta Professor Mota Pinto
3000-075 Coimbra
E-mail: gicalado@sapo.pt
Financiamento: Nenhum
Declaração de conflitos de interesse: Nenhum a Declarar
Data de recepção / Received in: 20/09/2011
Data de aceitação / Accepted for publication in: 04/03/2011

