










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Imunoalergologia
versão impressa ISSN 0871-9721
Rev Port Imunoalergologia vol.24 no.2 Lisboa jun. 2016
ARTIGO DE ATUALIZAÇÃO
Mecanismos imunopatológicos das reações de hipersensibilidade a fármacos
Immunopathological mechanisms of drug hypersensitivity reactions
Frederico Regateiro1,2, Emília Faria1
1 Serviço de Imunoalergologia, Centro Hospitalar Universitário de Coimbra, Portugal 2 Instituto de Imunologia, Faculdade de Medicina, Universidade de Coimbra, Portugal
RESUMO
As reações de hipersensibilidade (HS) imunológica a fármacos são mediadas pelos linfócitos do sistema imunitário adaptativo, apresentando especificidade e memória imunológica. A ativação de linfócitos requer reconhecimento específico do antigénio pelos recetores de linfócitos (sinal 1) e expressão de moléculas de coestimulação (sinal 2) pela célula apresentadora de antigénio (APC). O tipo de resposta inflamatória é determinado pelas citocinas existentes no microambiente da ativação (sinal 3). A origem destes três sinais nas HS a fármacos apresenta algumas particularidades.
O reconhecimento de pequenos fármacos depende de mecanismos de haptenização, pro‑haptenizacao ou p‑i concept.
Os sinais de coestimulação podem ser iniciados pela ação direta do fármaco na APC ou ter origem no microambiente.
As citocinas polarizam a resposta efetora dos linfócitos, dando origem a tipos de HS com manifestações clínicas diversas. O reconhecimento dos mecanismos imunopatológicos envolvidos é essencial para o diagnóstico, tratamento e orientação dos doentes com HS a fármacos.
Palavras‑chave: Alergia a fármacos, haptenos, hipersensibilidade a fármacos, p‑i concept, pro‑haptenos, reacções adversas a fármacos.
ABSTRACT
Immune‑mediateddrug hypersensitivity (HS) reactions involve lymphocyte activation and present the hallmark characteristics of adaptive responses: specificity and immunological memory. Lymphocyte activation requires specific recognition of the drug by lymphocyte receptors (signal 1) and co‑stimulation (signal 2) by the antigen presenting cell (APC). The type of inflammatory reaction is determined by the cytokine microenvironment during activation (signal 3). The origin of these stimulatory signals in drug HS is controversial. Small drug recognition by lymphocytes occurs by haptenization, pro‑haptenization and p‑I concept mechanisms. Co‑stimulatory signals might be induced on the APC by the drug itself or originate from the microenvironment surrounding the APC. Cytokines polarize the effector response of lymphocytes and give rise to different types of HS with distinct clinical manifestations. The recognition of the immunopathological mechanisms involved in the HS reactions is crucial for the diagnostics, treatment and management of patients with drug HS.
Key‑words: Adverse drug reaction, drug allergy, drug hypersensitivity, haptens, p‑I concept, prohaptens.
INTRODUÇÃO
Em 1972, a Organização Mundial de Saúde (OMS) definiu reacção adversa a medicamento como uma resposta nociva e indesejada a um fármaco que ocorre com doses normalmente utilizadas para profilaxia, diagnóstico ou terapêutica de doenças, ou para alteração de uma função fisiológica1. As reações adversas a medicamentos (RAM) ocorrem em cerca de 10 % dos doentes hospitalizados e em 7 % dos tratamentos ambulatórios2, causam morbilidade e mortalidade significativas e têm um impacto importante na prática clínica e na saúde pública.
As RAM dividem‑se, segundo a classificação proposta por Rawlins e Thompson3, em reações do tipo A e do tipo B. As RAMs do tipo A são reações previsíveis pelas propriedades farmacológicas ou toxicológicas do medicamento, dependentes da dose e que podem ocorrer em qualquer indivíduo quando exposto a uma dose suficiente do fármaco (embora a predisposição genética do indivíduo possa contribuir para a intensidade ou tipo da reação). Constituem cerca de 85 a 90 % de todas as RAMs e podem subdividir‑se em efeitos secundários, overdose e interações medicamentosas. As RAMs do tipo B, que constituem 10‑15% das reações adversas a fármacos, são reações que não dependem do efeito farmacológico do medicamento e que surgem apenas em indivíduos suscetíveis.
Podem dividir‑se em intolerâncias, idiossincrasias, reações não imunológicas e reações imunológicas.
Neste artigo, debrucamo‑nos sobre os mecanismos imunopatológicos envolvidos neste último tipo de reacções do tipo B: as reações de hipersensibilidade imunológica a fármacos. Classicamente estas reações foram descritas como reações imprevisíveis e independentes da dose, tendo sido por isso consideradas Bizarras (para colmatar falhas da dicotomia A/B, uma classificação mais recente das reações adversas divide as RAMs em tipos4: A – Aumentadas, B – Bizarras, C – Crónicas ou Cumulativas, D – do inglês Delayed, ou seja, efeitos a longo prazo, E – Eventos na suspensão do fármaco, e F – Falha terapêutica; para os dois primeiros tipos, A e B, a caracterização fisiopatológica é sobreponível aos tipos A e B classicamente descritos). As últimas décadas trouxeram avanços significativos na compreensão dos mecanismos imunopatológicos implicados e alguma da sua imprevisibilidade e independência da dose foi desmistificada. Como será descrito nesta revisão, é atualmente possível identificar, através de genotipagem, alguns dos doentes com risco aumentado de reação de hipersensibilidade (HS) a determinados fármacos. Por outro lado, a dose de fármaco administrada é claramente importante em praticamente todos os tipos de HS e, mesmo em reações de HS imediata dependentes de IgE, a dose tem um papel determinante, como fica demonstrado pelos protocolos de dessensibilização a fármacos em que doses pequenas são toleradas por indivíduos confirmadamente alérgicos.
As reações de hipersensibilidade imunológica a medicamentos apresentam características das respostas imunes adaptativas: especificidade e memória imunológica.
No entanto, os mecanismos imunopatológicos das reacções de hipersensibilidade a fármacos desafiam frequentemente o paradigma atual da iniciação de respostas adaptativas (sinais 1, 2 e 3 de ativação). Neste artigo analisamos as particularidades das reações de hipersensibilidade imunológica a fármacos, nomeadamente o reconhecimento específico dos fármacos pelos receptores dos linfócitos T, a coestimulação de linfócitos específicos de fármaco e os tipos de reações de hipersensibilidade a fármacos de acordo com a classificação mecanística de Gell e Coombs modificada.
A ATIVAÇÃO DO SISTEMA ADAPTATIVO NAS REAÇÕES A FÁRMACOS
O sistema imunitário adaptativo é constituído pelos linfócitos T e B (os linfócitos NK não possuem receptores específicos nem memória imunológica e são, regra geral, considerados parte da imunidade inata). Não é claro qual o papel, a existir, dos linfócitos B1 e dos linfócitos T γδ (apenas um clone específico de fármaco identificado5) nas reações de HS a fármacos, pelo que nos referiremos apenas aos linfócitos B2 e aos linfócitos T αβ e suas funções nas respostas específicas a fármacos.
As respostas adaptativas a fármacos incluem uma resposta primária (ou fase de sensibilização) e uma resposta secundária (ou fase de exposição/provocação).
Durante a sensibilização ocorre uma ativação e proliferação clonal de linfócitos T específicos para o fármaco que podem diferenciar‑se em células efetoras de HS ou estimular a resposta de linfócitos B e anticorpos. Na fase de exposição, mecanismos efetores dos diversos tipos de HS entram em ação causando doença imediata ou tardia. Os tipos de respostas efetoras serão descritos abaixo. Regra geral, a fase de sensibilização é assintomática e apenas na reexposição, ou na exposição prolongada, surgem os sinais e sintomas de HS. Como veremos adiante, nas reações a fármacos nem sempre este dogma da resposta adaptativa se verifica e, em alguns casos, os sintomas surgem numa primeira exposição ao fármaco.
A importância fisiopatológica da interação célula apresentadora de antigénio (APC) – antigénio (Ag) – receptor de linfócitos T (TCR) é ilustrada pelo número de Prémios Nobel da Fisiologia e Medicina (quatro) atribuídos a contribuições para a sua compreensão: 1980 – Baruj Benacerraf e Jean Dausset, descoberta do complexo major de histocompatibilidade (MHC); 1984 – Niels Jerne, teoria da seleção clonal, iniciada por MacFarlane Burnet (também ele Prémio Nobel em 1960); 1996 – Peter Doherty e Rolf Zinkernagel, interação MHC‑linfócitos T citotóxicos na resposta a vírus; 2011 – Ralph M. Steinman, células dendríticas, Bruce A. Beutler e Jules A. Hoffmann, toll‑like receptors (TLRs) e maturação de células dendríticas (DCs).
A resposta de linfócitos T é estimulada quando APCs, e em particular DCs, apresentam na sua superfície moléculas de MHC carregadas com peptídeos para os quais os linfócitos possuem TCRs específicos (sinal 1 de ativação, determina a especificidade da resposta) na presença de moléculas de co‑estimulação (e.g., CD80/86) que induzem a proliferação clonal dos linfócitos T (sinal 2). As moléculas de MHC da classe I apresentam peptídeos de origem intracelular com 8‑10 aminoácidos a linfócitos T CD8+ e as moléculas de MHC da classe II apresentam peptídeos de origem extracelular com 12‑16 Aminoácidos a linfócitos T CD4+ (excetua‑se a cross‑presentation de antigénios, um tema irrelevante para o entendimento deste artigo).
As citocinas produzidas pelas APCs e outras citocinas existentes no microambiente em que se dá a interacção APC‑linfocito T (sinal 3) determinam a polarização dos linfócitos (Th1, Th2, Th17, Tr1, etc.) e a sua função de modo a responderem adequadamente aos diferentes desafios patogénicos (e.g., linfócitos T CD4+ Th1, linfócitos T reguladores, etc.).
A interação entre linfócitos T CD4+ ativados e linfócitos B que apresentam o seu antigénio específico induz a proliferação clonal de linfócitos B e produção de anticorpos secundários específicos, obedecendo a sinais análogos de especificidade, coestimulação e diversificação funcional (e.g., mudança de classe de imunoglobulinas).
A forma como os fármacos desencadeiam esta sequência de ativação da resposta adaptativa, sinal 1, 2 e 3, é uma das mais importantes e enigmáticas questões na compreensão das reações de HS a fármacos. Como é que pequenos fármacos são reconhecidos pelos recetores de linfócitos? Quais são os sinais de perigo associados? Quais os mediadores do sinal 3 na resposta a fármacos que determinam o tipo de HS e patologia?
SINAL 1 – RECONHECIMENTO ESPECÍFICO DE FÁRMACOS PELOS LINFÓCITOS T
Os fármacos são moléculas quimicamente muito diversas, incluindo grandes proteínas multiméricas ou pequenas moléculas com apenas alguns iões (Quadro 1). Esta diversidade tem implicações no reconhecimento de cada tipo de fármaco pelas células do sistema imunitário.
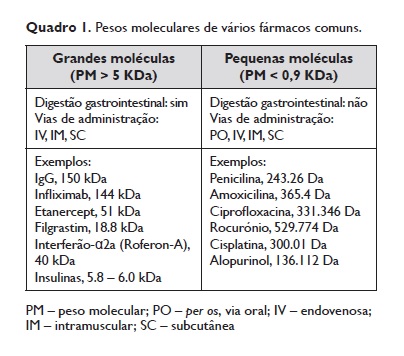
O reconhecimento de grandes moléculas obedece aos princípios habituais de antigenicidade: são antigénios completos apresentando múltiplos epitopos diferentes ou epitopos repetitivos e iniciam respostas específicas de forma clássica ou através do crosslinking de recetores. Tal está demonstrado em medicamentos como os anticorpos monoclonais terapêuticos6,7 ou os relaxantes neuromusculares (RMN), em que a presença de epitopos de amónio quaternário torna a succinilcolina bivalente e outros RNM multivalentes8.
A maioria dos fármacos, no entanto, são pequenas moléculas, uma característica essencial para que, por exemplo, se difundam através das membranas celulares atingindo alvos intracelulares ou para absorção e biodisponibilidade quando administradas pela via oral. Alguns fármacos têm pesos moleculares inferiores ao de simples aminoácidos (os aminoácidos têm pesos moleculares entre os 75Da da Glicina e os 204Da do Triptofano).
A título de exemplo, a penicilina é pouco maior do que o triptofano, enquanto o alopurinol é menor do que alguns aminoácidos (Quadro 1). Tendo em conta que as moléculas de MHC classe I apresentam peptídeos com 8 a 10 aminoácidos (~900‑1100Da) ao TCR/CD8+ e que o MHC classe II apresenta peptídeos com 12 a 16 aminoácidos (~1300‑1800Da) ao TCR/CD4+, a forma como estas pequenas moléculas são reconhecidas pelos recetores do sistema imunitário não é óbvia e tem sido alvo de curiosidade científica desde há décadas9. Vários modelos têm sido propostos para explicar o reconhecimento de pequenos fármacos pelos recetores específicos dos linfócitos, dos quais destacaremos, pelo nível de evidência, os modelos de haptenização, pro‑haptenizacao e o p‑i 3concept.
Haptenização
Neste modelo, a formação de complexos, através de ligações covalentes entre proteínas autólogas e uma molécula de baixo peso molecular isoladamente não imunogénica (como um pequeno fármaco), cria neoantigénios com epitopos que podem ser reconhecidos pelos receptores do sistema imunitário. A possibilidade de formar complexos de haptenização depende da reatividade do fármaco com as proteínas autólogas, um fenómeno que pode ser previsto e que tem importância no desenho de novos fármacos. O conceito de haptenização foi proposto há mais de oito décadas por Landsteiner e Jacobs em 19359, ao identificarem a associação entre a reatividade com proteínas de um composto e o seu potencial imunogénico, citando trabalhos já anteriores de R.L. Mayer (1928) e L. Dienes (1933) sobre a reatividade da p‑fenilenediamina com proteínas.
A haptenização foi demonstrada em vários fenómenos biológicos e em algumas patologias, como a dermatite de contacto alérgica ao níquel, e está também identificado em várias HS a fármacos que formam ligações covalentes com proteínas celulares ou do plasma (e.g., albumina, integrinas, enzimas, etc.). A importância da haptenização está bem estabelecida nas reações de hipersensibilidade a antibióticos β‑lactamicos10,11, sendo inclusivamente conhecidos os exatos resíduos de lisina da albumina sérica em que ocorre a interação fármaco/proteína12. A haptenização está também demonstrada para reações de HS a cefalosporinas, carbapenem, monobactam, penicilamina e hidralazina, entre outros.
Pro‑haptenizacao
O conceito de pro‑haptenizacao é semelhante ao de haptenização (ligação covalente a proteínas e formação de neoantigénios), mas é precedido da metabolização do fármaco. Muitos fármacos são quimicamente inertes na sua forma original e, na sua forma nativa, não têm capacidade de se ligar a proteínas. Para que se tornem quimicamente (proteína) reativos têm que ser bioativados pelos processos normais de metabolismo – regra geral originando metabolitos eletrofílicos que se ligam a resíduos nucleofílicos nas proteínas.
Uma implicação que decorre da necessidade de bioativação é a de que a imunogenicidade e as manifestações clínicas podem restringir-se aos locais onde ocorre a metabolização do medicamento (e.g., hepatite ou nefrite), o que pode explicar sintomas específicos de órgão ou tecido que ocorrem em algumas HS a fármacos.
O protótipo das HS por pro‑haptenizacao é o sulfametoxazol, uma pequena molécula de 253 Da que não reage com proteínas na forma original mas que, ao ser metabolizada no fígado pelo citocromo P450 em sulfametoxazol hidroxilamina e oxidado em sulfametoxazol nitroso, ganha capacidade de se ligar a proteínas celulares e séricas, modificando grupos tiol e produzindo novos epitopos13. Cerca de 90 % dos linfócitos T envolvidos em reações de HS a sulfametoxazol são dependentes da sua metabolização prévia e são específicos para modificações de proteínas dependentes da produção de sulfametoxazol nitroso14. Outros exemplos de reconhecimento imune de fármacos por pro‑haptenizacao incluem reações de HS a sulfonamidas, halotano, fenitoína, carbamazepina, lamotrigina, entre outros.
p‑i concept
Os fenómenos de haptenização e pro‑haptenizacao não foram demonstrados para a generalidade dos fármacos e não explicam a existência de reações a fármacos quimicamente não reativos ou não metabolizados. Em 2002, Werner J. Pichler propôs um novo modelo de reconhecimento específico a que chamou p‑i concept15 (de Pharmacologic interaction of drugs with Immune receptors, expressão ainda sem equivalente estabelecida na língua portuguesa). Segundo o modelo de p‑i, os fármacos não funcionam como antigénios, mas causam reações de HS ao ligarem‑se diretamente ao TCR ou ao HLA, induzindo a ativação e a proliferação clonal de linfócitos T após contacto com a APC (o p‑i concept não foi ainda demonstrado em reações por linfócitos B/anticorpos ou NK). Os recetores de linfócitos (no indivíduo), bem como as moléculas de HLA (na população), são altamente variáveis na sua conformação e zonas de ligação ao antigénio (mais de 1011 TCRs diferentes podem ocorrer num indivíduo e mais de 9900 alelos de HLA classe I e mais de 3000 classe II estão descritos na população humana – http://www.allelefrequencies.net/default.asp), tornando provável que algumas destas moléculas tenham zonas de ligação semelhantes às dos alvos para os quais os fármacos foram desenhados. Pode, assim, formar‑se uma ligação ao receptor imune semelhante à ligação do fármaco ao seu ligando terapêutico (não covalente, por forças de van der Waals, eletroestáticas ou pontes de hidrogénio)13. Segundo este modelo, a ativação dos linfócitos não decorre da estimulação imunológica por antigénios, mas por estimulação farmacológica de um recetor de linfócitos pelo fármaco.
Linfócitos T de memória ou pre‑ativados (e.g., por infeções) serão particularmente sensíveis a este tipo de estimulação por apresentarem um limiar de ativação mais baixo. A especificidade dos linfócitos T ativados por p‑i é geralmente desconhecida e pode ter importantes implicações nas manifestações clínicas.
O p‑i concept permite explicar algumas observações clínicas e laboratoriais que não são compatíveis com o modelo hapteno/pro‑hapteno13,16: 1. os fármacos nem sempre se ligam de forma covalente às proteínas, ligando‑se, frequentemente, de forma lábil através de ligações não covalentes; 2. células dendríticas fixadas, ou seja, incapazes de processar antigénios ou de metabolizar pro‑haptenos, podem ativar respostas T específicas quando incubadas com fármacos quimicamente inertes17; 3. A maioria das reações de HS a fármacos é mediada apenas por linfócitos T, não existindo uma resposta completa com linfócitos T, linfócitos B e anticorpos, como pode acontecer com antigénios completos, como os haptenizados; 4. ocorrem reações muito rápidas, praticamente imediatas, mediadas por linfócitos T, sem a presença de anticorpos; 5. é possível estimular diretamente clones de células T de forma reversível e dependente da dose; 6. reações de HS podem ocorrer sem contacto prévio, na primeira administração, sem fase de sensibilização; 7. Algumas reações de HS são previsíveis e fortemente associadas a alelos ou haplótipos HLA.
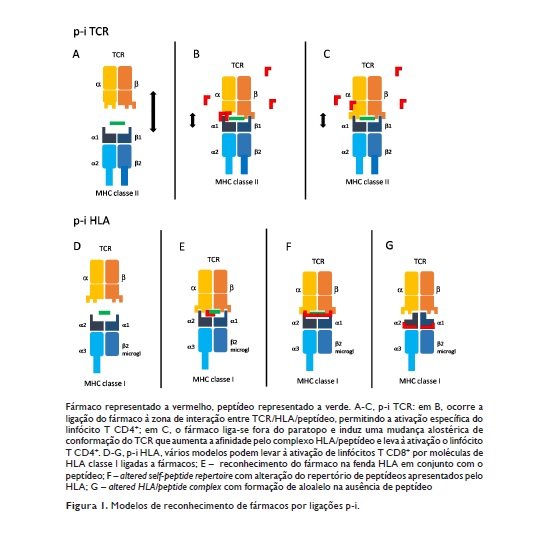
Segundo este modelo, a estimulação de clones de linfócitos T pode acontecer por ligação do fármaco directamente ao TCR5,18 (p‑i TCR) ou por ligação do fármaco ao HLA19,20 (p‑i HLA)(Figura 1)21.
p‑i TCR
O p‑i TCR parece ser mais frequente em respostas de linfócitos T CD4+ e e stá e nvolvido e m e xantemas maculopapulares e, raramente, em situações mais graves como o DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms, também designado por DiHS, Drug‑induced Hypersensitivity Syndrome). No p‑i TCR, a ativação dos linfócitos T ocorre por ligação do fármaco ao TCR, sendo também necessária a interação do TCR com complexos HLA/peptídeo. Significativamente, não existe restrição HLA, podendo ocorrer com HLAs alogénicos22, ou mesmo removendo ou alterando o peptídeo da fenda do HLA23. O fármaco pode ligar‑se ao TCR na zona de interação/ligação ao HLA (Figura 1.B) ou a regiões do TCR fora da interação com o HLA, mas induzindo modificações alostéricas da molécula de TCR e alterando a sua afinidade para o HLA/peptídeo24,25 (Figura 1.C). Alguns clonotipos de TCR envolvidos no reconhecimento de fármacos foram recentemente identificados26. No entanto, regra geral, não se conhecem as especificidades dos TCRs a que se ligam os fármacos causadores de HS. Os mesmos fármacos tendem a induzir fenótipos clínicos semelhantes em vários indivíduos, pelo que é possível que cada medicamento tenda a ativar especificidades semelhantes de TCR27.
p‑i HLA
O p‑i HLA estimula respostas sobretudo de linfócitos T CD8+ e pode estar na origem de reações citotóxicas graves, como a Síndrome de Stevens‑Johnson (SJS) ou a Necrólise Epidérmica Tóxica (TEN). Nestes casos, existe uma forte restrição HLA, uma vez que é necessário que exista ligação direta do fármaco às moléculas de HLA19,20.
Esta associação entre alelos/haplótipos HLA e reacções de HS a alguns fármacos foi inicialmente descrita em 200228,29 (associação entre alelos HLA e a suscetibilidade para reacções de HS ao abacavir em indivíduos HIV+) e tem importantes implicações clínicas, uma vez que permite prever (e evitar) a ocorrência de reações adversas graves a alguns fármacos através da genotipagem prévia à administração do fármaco.
Regra geral, as associações HLA/HS a fármaco são específicas de fármaco e apresentam elevados valores preditivos negativos. No entanto, os valores preditivos positivos (VPP) são baixos (possivelmente limitados pela presença de clonotipos TCR para o fármaco). Uma excepção a esta regra é a associação entre HLA‑B* 57:01 e HS grave a abacavir, que apresenta um VPP de 47 % e um risco relativo de 100028,29, pelo que, atualmente, está generalizado o rastreio de HLA pre‑tratamento com este fármaco30.
Existem publicadas31 numerosas associações HLA/HS a fármacos específicos, algumas delas apenas em populações selecionadas. A associação entre HLA‑B* 58:01 e reações adversas cutâneas graves, descrita sobretudo em chineses da etnia Han, foi recentemente confirmada numa população portuguesa de doentes com SJS/TEN e DRESS32.
No p‑i HLA, a ligação do fármaco ao HLA pode ocorrer por via intracelular (ligação ao HLA ainda vazio no retículo endoplasmático da APC – a que se segue a ligação do HLA aos seus peptídeos habituais, mas também a peptídeos para os quais, pela presença do fármaco, ganha uma nova afinidade, originando um altered self‑peptide repertoire19,20) ou por via extracelular (ligação do fármaco ao HLA já expresso na superfície da APC e carregado com peptídeo).
A estimulação do TCR pelo complexo fármaco/HLA/peptídeo pode ocorrer de, pelo menos, três formas diferentes (Figura 1): 1. Reconhecimento do fármaco apresentado na fenda do HLA em conjunto com o HLA/peptídeo (Figura 1.E) –, como parece ser o caso do reconhecimento da carbamazepina ligada ao HLA‑B* 15:0233 – ou, eventualmente, mesmo sem peptídeo na fenda HLA27; 2. altered self peptide repertoire (Figura 1.F), em que o fármaco ligado ao HLA altera o repertório de peptídeos endógenos que se podem ligar e ser apresentados pelo HLA, induzindo uma alteração do self imunológico e causando respostas auto‑imunes policlonais – como parece ocorrer para a associação HLA‑B* 57:01 e abacavir19,20,34; 3. altered HLA/peptide complex (Figura 1.G), em que a ligação do fármaco ao HLA altera a conformação da fenda e transforma o HLA num aloalelo contra o qual numerosos clones de linfócitos T naïve ou de memória reagem imediatamente (à semelhança da rejeição de transplantes alogénicos), mesmo na ausência de peptídeo na fenda HLA35.
Outros modelos
Modelos menos consensuais incluem o heterologous immune model, que postula que a HS a fármacos é desencadeada por reatividade cruzada de linfócitos T de memória dirigidos a infeções virais frequentes, como os herpes vírus humanos (e.g., HHV‑6)36.
Finalmente, é de notar que os vários modelos não são mutuamente exclusivos e certos fármacos podem ser reconhecidos tanto por haptenização como por mecanismos p‑i (p‑i TCR e/ou p‑i HLA), com manifestações clínicas totalmente distintas. Tal foi observado, por exemplo, para a flucloxacilina, que tanto pode ser reconhecida por p‑i e causar hepatite tóxica (drug‑induced liver injury, DILI) em doentes com HLA‑B* 57:0137, como por mecanismos dependentes de haptenização em indivíduos sem este alelo38.
SINAL 2 (DANGER SIGNAL) NA ACTIVAÇÃO DE LINFÓCITOS T ESPECÍFICOS
O sinal 2 de ativação dos linfócitos T é iniciado pelo reconhecimento de sinais de perigo (segundo o danger model proposto por Polly Matzinger39) pelas células dendríticas.
Os recetores PRR (pattern recognition receptors) existentes nas células do sistema inato e adaptativo reconhecem moléculas com padrões conservados filogeneticamente associados a patogénios (PAMPs, pathogen associated molecular patterns, por exemplo, ADN viral ou lipopolissacarídeo bacteriano), ou a perigo para o organismo (DAMPs, danger associated molecular patterns, por exemplo, sinais de necrose como o ATP extracelular, etc.). Quando ativados, os PRR levam à maturação da célula dendrítica, promovendo a sua migração para os gânglios linfáticos, a extensão de dendrites, e aumentando a expressão de HLA na superfície da membrana celular e de um conjunto de moléculas de adesão e de coestimulação que vão ativar os linfócitos específicos para o HLA/peptídeo e induzir a sua proliferação, polarização e produção de citocinas.
A forma como os fármacos induzem a maturação das células do sistema inato é controversa. A generalidade dos fármacos não se liga diretamente a PRR e é administrada de formas inócuas sem aparentes sinais de perigo associados, por exemplo através da via oral. Se, por um lado, o uso de medicação pressupõe uma doença subjacente e algumas patologias envolvem sinais de perigo reconhecidos (e.g., antibióticos e anti‑inflamatorios nas infeções, citostáticos no cancro, ou durante trauma cirúrgico), existem também múltiplas situações em que nem a doença nem a administração do fármaco implicam sinais de perigo evidentes, mas ocorrem reações de HS (e.g., anti‑hipertensores, anestésicos, contrastes, etc).
Para além disso, há situações com evidentes sinais de perigo que não originam reações de HS (e.g., paracetamol em doses elevadas causa necrose hepática não imune mediada, uma situação com abundantes sinais de perigo e com possibilidade de haptenização demonstrada, mas que não inicia respostas imunes).
A maturação das DCs e coestimulação associados a HS a fármacos parece ocorrer por vários mecanismos com origem no próprio fármaco ou no ambiente que rodeia a DC:
Ligação direta do fármaco a PRR, um mecanismo conhecido dos agonistas do TLR7/8 imiquimod e outras imidazoquinolinas40;
Ligação covalente de metabolitos reativos do fármaco a ADN ou a proteínas importantes para a sobrevivência ou ativação da DC, comprometendo funções celulares essenciais e causando stress na DC. Tal efeito foi demonstrado para a amoxicilina e o sulfametoxazol41. Rodriguez‑Pena e colegas42 demonstraram que a amoxicilina induz a maturação das células dendríticas apenas de doentes alérgicos a este fármaco, mas não em células dendríticas de controlos não alérgicos. Os mecanismos envolvidos não foram ainda esclarecidos;
Produção de intermediários reativos e tóxicos após metabolização do fármaco pela própria DC, por exemplo a metabolização celular do SMX em SMX‑NO produz radicais livres de oxigénio, stress oxidativo e maturação da DC43;
Efeitos tóxicos do medicamento em células na vizinhança da DC com libertação de sinais de perigo que são reconhecidos pelos PRR da célula dendrítica;
Outros sinais associados à doença e não ao fármaco, podendo ser internos à DC (stress químico, físico, infecioso, inflamatório, oxidativo) ou externos à DC (infecioso, trauma, etc.)44.
Estes sinais podem ocorrer isoladamente ou em combinação. Por exemplo, a exposição de APCs a sinais de perigo conhecidos (lipopolissacarídeo bacteriano, enterotoxina B estafilocócica, IL‑6, IFN‑γ, etc.) não só induz ativação como também aumenta a produção de aductos reativos do SMX tóxicos para a DC44.
Algumas HS a fármacos são mais frequentes em determinadas doenças, por exemplo existe uma associação entre fibrose quística e HS a antibióticos45 e um risco aumentado de HS a sulfametoxazol em doentes HIV/SIDA, em particular se existir replicação viral ativa46,47. Quanto destas incidências é atribuível à maior utilização do fármaco nestas patologias e quanto será atribuível a mecanismos de coestimulação por interação infeção/fármaco/sistema imunitário não está completamente esclarecido.
São também conhecidas as maiores incidências de rash medicamentoso associadas a infeções virais nas crianças e os fenómenos de reativação de infeções crónicas por herpes vírus (HHV 6, HHV 7, CMV e EBV) no DRESS/DiHS/síndrome de hipersensibilidade48 com recrudescência dos sintomas de HS ao fármaco durante os períodos de reativação viral36. Estas reações de HS podem ocorrer com vários fármacos e os mecanismos responsáveis por estas associações não são totalmente compreendidos49.
No entanto, para além da co‑estimulacao, a infeção ou outras patologias podem desencadear mecanismos imunológicos (como a pre‑ativacao de linfócitos ou a reatividade cruzada) ou não imunológicos (como alterações da farmacocinética e metabolização do fármaco, interacções medicamentosas, etc.) que levem a susceptibilidade aumentada a HS a fármacos50. A forma como uma determinada infeção em particular induz suscetibilidade a um fármaco específico permanece por esclarecer e, provavelmente, terá de ser estudada individualmente para cada combinação infeção/fármaco.
Finalmente, é possível, pelo menos no plano teórico, que algumas HS por p‑i HLA possam dispensar coestimulação, atuando por reconhecimento de alo‑HLA ou em situações que envolvam linfócitos T de memória ou linfócitos T pre‑ativados por infeções ou episódios prévios de HS ao fármaco21.
MECANISMOS EFETORES – TIPOS DE HIPERSENSIBILIDADE A FÁRMACOS
Diversas formas de sistematização das reações de HS foram propostas por diversos autores, mas aquela que parece reunir maior consenso é a classificação de Gell e Coombs. Em 1963, os imunologistas Philip Gell e Robin Coombs propuseram uma classificação das reações de hipersensibilidade em quatro tipos, de acordo com os seus mecanismos imunopatológicos. Em 1968, os mesmos imunologistas aplicaram esta classificação para caracterizar as reações de hipersensibilidade a fármacos51. Apesar de sucessivamente modificada por vários autores, esta classificação mantem‑se útil para a compreensão dos mediadores implicados nas reações de HS a fármacos, contribuindo para a decisão diagnóstica, terapêutica e de prognóstico destes doentes. A classificação de Gell e Coombs modificada inclui 4 tipos de hipersensibilidade(Quadro 2):
Hipersensibidade de tipo I, também designada imediata ou alérgica, ocorre após ligação do antigénio (geralmente solúvel, de grandes dimensões ou haptenizado) a anticorpos específicos da classe IgE localizados na superfície de mastócitos e basófilos induzindo a sua desgranulação e libertação de mediadores pré‑formados, causadores dos sintomas imediatos (e.g., histamina, triptase, etc.), ou formados de novo (como leucotrienos, TNF‑α, etc.), de que resultam os sintomas tardios. A consequência desta rápida libertação de mediadores vasoativos são as manifestações clínicas típicas da doença alérgica (congestão nasal e ocular, obstrução nasal, broncospasmo, urticária, angioedema, anafilaxia ou choque anafilático), mas também outros, como o aumento da produção de muco, aumento da peristalse, etc.
A produção de IgE específica para o fármaco ocorre após um contacto prévio com o fármaco geralmente haptenizado a proteínas e desenvolvimento de plasmócitos produtores de IgE específica que se liga a recetores Fcε nos mastócitos/basófilos (fase de sensibilização). Após a fase de sensibilização e produção da resposta secundária IgE, o contacto subsequente com o antigénio desencadeia reacções rápidas ou imediatas, uma vez que tanto os anticorpos específicos como os mediadores vasoactivos dos grânulos dos mastócitos/basófilos estão pré-formados e disponíveis para iniciar a reação. Os sintomas clínicos, no entanto, podem demorar até mais de uma hora a surgir, em particular no caso de administração oral do fármaco, em que a absorção pode ser mais demorada e atrasada pela ingestão concomitante de alimentos.
Recentemente, foi notado que, numa grande proporção de doentes com reações imediatas a fármacos, não havia história de um contacto prévio com o fármaco que pudesse ter induzido a sensibilização – alguns autores estimam que em cerca de 50 % das HS imediatas não é possível identificar um contacto prévio com o fármaco52, o que sobe para 80% nas anafilaxias fatais53. Excluindo a possibilidade de ter havido uma toma desconhecida ou entretanto esquecida pelo doente ou família, uma possível explicação é de que tenha ocorrido sensibilização silenciosa a uma molécula para a qual exista reatividade cruzada com o fármaco. Um exemplo recentemente identificado é o da anafilaxia após a primeira administração de cetuximab, um anticorpo monoclonal terapêutico contra o epidermal growth factor receptor (EGFR) usado no tratamento do cancro colorrectal7. Estes doentes apresentam IgE anti‑cetuximab, em particular anti‑galactose‑α‑1,3‑galactose, antes de iniciar tratamento7, tendo sido sensibilizados para o epitopo através de picada de carraça54 (um exemplo de reatividade cruzada que pode originar também episódios de anafilaxia retardada após ingestão de carnes vermelhas).
Hipersensibilidade de tipo II, também designada citotóxica, é mediada por anticorpos das classes IgG ou IgM dirigidos a moléculas localizadas na superfície de células que, desta forma, se tornam alvos para células com capacidade citotóxica (como os linfócitos NK), células fagocíticas (em particular do sistema fagocítico mononuclear) ou para a lise celular por fixação de complemento e formação do complexo de ataque à membrana. Alguns fármacos ligam‑se a moléculas na superfície de células, como os eritrócitos ou plaquetas, e tornam‑se alvos para anticorpos IgG antifármaco que vão levar à destruição da célula, causando anemia hemolítica ou trombocitopenia, entre outros.
Hipersensibilidade de tipo III, também designada hipersensibilidade por imunocomplexos, ocorre pela ligação de anticorpos da classe IgG (e raramente IgM) a antigénios solúveis formando imunocomplexos circulantes que, se em grande quantidade, se podem depositar no endotélio vascular de alguns tecidos, membranas sinoviais ou membrana basal glomerular. Os imunocomplexos em excesso causam inflamação (e.g., vasculite) e dano de tecidos e órgãos por dois mecanismos principais: 1. Reconhecimento por recetores Fc de neutrófilos, macrófagos, mastócitos e outros leucócitos, levando à sua ativação; 2. ativação da via clássica do complemento e produção de C5a.
Hipersensibilidades do tipo IV, também designadas tardias, retardadas ou celulares, incluem respostas mediadas por vários tipos de linfócitos T.
Geralmente são tardias, após vários dias de exposição, mas podem ser rápidas, em apenas algumas horas se mecanismos p‑i estiverem envolvidos.
Atingem frequentemente a pele (um órgão rico em linfócitos T e APCs) e podem originar sintomatologia grave e fatal, contribuindo, no seu conjunto, para mais mortes do que as reações anafiláticas a fármacos. De acordo com o tipo de citocinas produzidas pelos linfócitos T e com o tipo de células efetoras, as reações de tipo IV dividem‑se em:
– IVa – mediadas por células com fenótipo Th1 produtoras de IFN‑γ e TNF‑α, com ativação de monócitos/macrófagos. Os linfócitos Th1 têm também capacidade de ativar linfócitos T CD8+, levando à combinação frequente da HS IVa com mecanismos da HS IVc, como ocorre na dermatite de contacto alérgica;
– IVb – mediada por células Th2 produtoras de IL4/IL5/IL13 com recrutamento e ativação de eosinófilos. Podem causar reações graves, como o DRESS, e estão frequentemente associadas à fase crónica das patologias de HS de tipo I;
– IVc – são mediadas por linfócitos T CD4+ e CD8+ com capacidade citotóxica, podendo causar reacções ligeiras, como o exantema maculopapular, ou reações graves com destruição tecidular extensa, como a síndrome de Stevens‑Johnson (STS), a necrólise epidérmica tóxica (TEN), ou hepatites tóxicas. Neste tipo de HS os próprios linfócitos T são as células efetoras, através da expressão de FasL e da produção de granzimas, perforina e granulisina (uma molécula produzida por linfócitos T CD8+ e N K e r ecentemente identificada como a principal responsável pela morte generalizada dos queratinócitos no SJS ou NET55);
– IVd – mediadas por linfócitos produtores de CXCL‑8/GM‑CSF com recrutamento de neutrófilos originando reações pustulosas estéreis, como a pustulose exantemática generalizada aguda (AGEP).
As reações de hipersensibilidade a fármacos do tipo I e IV são mais frequentes do que as dos tipos II ou III.
As reações tipo II e tipo III dependem, geralmente, de grandes quantidades de antigénio e no caso das HS a fármaco ocorrem apenas em tratamentos prolongados com doses altas de medicamentos. No Quadro 2 agrupamos as HS I‑IV de acordo com alguns critérios adicionais que nos parecem úteis para a compreensão dos desencadeantes, dos mediadores e das suas manifestações clínicas:
As HS de tipo I, II e III são mediadas por anticorpos de diversas classes; as de tipo IV são mediadas por linfócitos T, de diferentes polarizações funcionais;
O reconhecimento específico de fármacos nas HS dos tipos I, II e III depende da haptenização/pro‑haptenizacao do fármaco; nas HS de tipo IV o reconhecimento pode ser do tipo haptenos/pro‑haptenos ou do tipo p‑i concept;
Nas HS do tipo I, III e IVd, pode haver reconhecimento de fármacos solúveis ou ligados a proteínas em solução, enquanto nas HS dos tipos II, IVa, IVb e IVc, os fármacos reconhecidos estão ligados à matriz extracelular ou à superfície das células, ou são por elas apresentados no contexto do MHC;
As reações do tipo I, imediatas, são regra geral mais rápidas do que as restantes reações, uma vez que todos os mediadores se encontram pre‑formados e prontos a serem libertados. No entanto, as reações de tipo IV, em que o reconhecimento ocorre por mecanismos p‑i, podem ser muito rápidas e desenvolver‑se em apenas alguns minutos.
A classificação de Gell e Coombs apresenta algumas insuficiências na caracterização das reações imunológicas a fármacos:
A classificação é uma simplificação de fenómenos complexos que ocorrem in vivo e envolvem muitas outras células e moléculas efetoras. Descreve bem a fisiopatologia das reações de tipo I, mas não é exaustiva a descrever outros tipos de HS;
Frequentemente, o quadro clínico apresenta características mistas de vários tipos de HS, por exemplo I/IVb nas reações imediatas com fase crónica eosinofílica, IVa/IVc nas dermatites de contacto alérgicas, IVb/IVc nos exantemas maculopapulares, etc.;
Não explica todas as patologias provocadas por HS a fármacos, por exemplo a erupção fixa a fármaco e as reações de autoimunidade induzida por fármacos: as reações lupus‑like (amiodarina, isoniazida, etc.), penfigo‑like (penicilamina) ou a dermatose bolhosa linear por IgA (vancomicina, ceftriaxone, ciprofloxacina, metronidazol) não parece encaixarem em nenhum dos modelos de HS de Gell e Coombs;
Algumas reações de HS a fármacos apresentam manifestações específicas de órgão, enquanto outras parecem ser reações sistémicas de hipersensibilidade.
A classificação de Gell e Coombs não inclui mecanismos fisiopatológicos específicos de tecido ou de órgão, nem eventuais mecanismos de tolerância imunológica envolvidos13,56.
A World Allergy Organization (WAO) recomenda a classificação das reações de hipersensibilidade a fármacos em duas categorias: imediatas (que ocorrem na 1.ª hora após a administração do fármaco) e tardias (que ocorrem após a 1.ª hora, mais frequentemente após mais de 6 horas). Esta classificação temporal tem algum significado na identificação de reações mediadas pela IgE com potencial para causar anafilaxia com risco de vida na readministração (os fármacos são a causa mais frequente de anafilaxia fatal nos EUA, sendo responsáveis por 1445 mortes em 11 anos57) mas é insuficiente para explicar outros mecanismos patológicos.
É nossa opinião que a classificação de Gell e Coombs continua válida e com utilidade clínica na maioria das reações: permite identificar o principal mecanismo imunopatológico envolvido, o que tem implicações na decisão terapêutica, na escolha dos exames complementares de diagnóstico e no estudo imunológico necessário para cada doente.
CONCLUSÃO
As reações de HS imunológicas a fármacos têm mecanismos imunopatológicos diversos e não devem ser consideradas uma patologia única. A identificação do mecanismo de HS envolvido tem implicações clínicas importantes no diagnóstico, tratamento, prognóstico e rastreio. Estas conclusões, aparentemente óbvias, podem ser facilmente esquecidas durante a prática clínica. Deve, ainda, ser tido em conta que o mesmo fármaco pode causar diferentes tipos de HS em indivíduos ou ocasiões diferentes.
É possível que a imprevisibilidade das reações de HS imunológica a fármacos traduza limitações do nosso conhecimento e não seja, como tem sido interpretado até agora, uma característica inerente a este tipo de reações.
Os estudos de associação entre alelos ou haplótipos HLA e reações de HS de tipo IV a fármacos vieram clarificar alguma desta imprevisibilidade. O rastreio da susceptibilidade genética para reações de HS constitui um exemplo notável de medicina personalizada. O elevado valor preditivo negativo destes testes permite administrar o medicamento com segurança ao identificar os indivíduos em risco e evitar reações graves ou fatais. Por outro lado, o valor preditivo positivo destas associações é geralmente baixo e permanecem dúvidas sobre que outros factores determinam a ocorrência de reações.
No futuro, é de esperar que avanços da imunologia, metabolismo ou farmacogenética permitam caracterizar totalmente o risco de um indivíduo desenvolver HS a um determinado fármaco. A forma como algumas infecções funcionam como cofatores das reações de HS, a manutenção a memória imunológica para o fármaco, os mecanismos envolvidos na dessensibilização a fármacos e a possibilidade de indução de tolerância duradoura a fármacos são outras das questões fundamentais da imunopatologia das HS a fármacos para as quais a resposta permanece incerta.
REFERÊNCIAS
1. World Health Organization. Internacional drug monitoring: the role of national centres. Report of a WHO Meeting. 1972: 1-25. [ Links ]
2. Gomes ER, Demoly P. Epidemiology of hypersensitivity drug reactions. Curr Opin Allergy Clin Immunol 2005;5:309‑16. [ Links ]
3. Rawlins M, Thompson J. Pathogenesis of adverse drug reactions. In: Textbook of adverse drug reactions. 1977:10. [ Links ]
4. Aronson J. Drug therapy. In: In: Haslett C, Chilvers ER, Boon NA, Colledge NR, Hunter JAA (Eds.). Davidsons principles and practice of medicine 19th ed Edinburgh: Elsevier Science. 2002:47‑63.
5. Zanni MP, Mauri‑Hellweg D, Brander C, Wendland T, Schnyder B, Frei E, et al. Characterization of lidocaine‑specific T cells. J Immunol 1997;158:1139‑48. [ Links ]
6. Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics The key causes, consequences and challenges. Self/Nonself 2010;1:314‑22. [ Links ]
7. Chung CH, Mirakhur B, Chan E, Le Q‑T, Berlin J, Morse M, et al. Cetuximab‑induced anaphylaxis and IgE specific for galactose‑alpha‑1,3‑galactose. N Engl J Med 2008;358:1109‑17. [ Links ]
8. Didier A, Cador D, Bongrand P, Furstoss R, Fourneron P, Senft M, et al. Role of the quaternary ammonium ion determinants in allergy to muscle relaxants. J Allergy Clin Immunol 1987;79:578‑84. [ Links ]
9. Landsteiner K, Jacobs J. Studies on the sensitization of animals with simple chemical compounds. J Exp Med 1935;61:643‑56. [ Links ]
10. Batchelor FR, Dewdney JM GD. Penicillin allergy: the formation of the penicilloyl determinant. Nature 1965;206:362‑4. [ Links ]
11. Brander C, Mauri‑Hellweg D, Bettens F, Rolli H, Goldman M PW. Heterogeneous T cell responses to beta‑lactam‑modified self‑structures are observed in penicillin‑allergic individuals. J Immunol 1995;155:2670‑8. [ Links ]
12. Jenkins RE, Meng X, Elliott VL, Kitteringham NR, Pirmohamed M PB. Characterisation of flucloxacillin and 5‑hydroxymethyl flucloxacillin haptenated HSA in vitro and in vivo. Proteomics Clin Appl 2009;3:720‑9. [ Links ]
13. Pichler WJ, Naisbitt DJ, Park BK. Immune pathomechanism of drug hypersensitivity reactions. J Allergy Clin Immunol 2010; 127:S74‑81. [ Links ]
14. Castrejon JL, Berry N, El‑Ghaiesh S, Gerber B, Pichler WJ, Park BK, et al. Stimulation of human T cells with sulfonamides and sulfonamide metabolites. J Allergy Clin Immunol 2010;125:411‑18. [ Links ]
15. Pichler W. Pharmacological interaction of drugs with antigen‑specific immune receptors: the p‑I concept. Curr Opin Allergy Clin Immunol 2002;2:301‑5. [ Links ]
16. Pichler WJ, Watkins S. Interaction of small molecules with specific immune receptors: The p‑I concept and its consequences. Current Immunology Reviews 2014;7‑18. [ Links ]
17. Zanni MP, Von Greyerz S, Schnyder B, Brander KA, Frutig K, Hari Y, et al. HLA restricted, processing‑and metabolism‑independent pathway of drug recognition by human α T lymphocytes. J Clin Invest 1998;102:1591‑8. [ Links ]
18. von Greyerz S, Zanni MP, Frutig K, Schnyder B, Burkhart C, Pichler WJ. Interaction of sulfonamide derivatives with the TCR of sulfamethoxazole‑specific human alpha beta+ T cell clones. J Immunol 1999;162:595‑602. [ Links ]
19. Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, et al. Immune self‑reactivity triggered by drug‑modified HLA‑peptide repertoire. Nature 2012; 486:554‑8. [ Links ]
20. Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, et al. Drug hypersensitivity caused by alteration of the MHC‑presented self‑peptide repertoire. Proc Natl Acad Sci USA 2012;109:9959‑64. [ Links ]
21. Adam J, Pichler WJ, Yerly D. Delayed drug hypersensitivity: Models of T‑cell stimulation. Br J Clin Pharmacol 2011;71:701‑7. [ Links ]
22. von Greyerz S, Bültemann G, Schnyder K, Burkhart C, Lotti B, Hari Y, et al. Degeneracy and additional alloreactivity of drug‑specific human alpha beta(+) T cell clones. Int Immunol 2001;13:877‑85. [ Links ]
23. Burkhart C, Britschgi M, Strasser I, Depta JPH, Von Greyerz S, Barnaba V, et al. Non‑covalent presentation of sulfamethoxazole to human CD4+ T cells is independent of distinct human leucocyte antigen‑bound peptides. Clin Exp Allergy 2002;32(11):1635‑43. [ Links ]
24. Watkins S, Pichler WJ. Activating interactions of sulfanilamides with T cell receptors. Open J Immunol 2013;3:139‑57. [ Links ]
25. Watkins S, Pichler WJ. Sulfamethoxazole induces a switch mechanism in T cell receptors containing TCRVβ20‑1, altering pHLA recognition. PLoS One 2013;8 e76211. [ Links ]
26. Ko TM, Chung WH, Wei CY, Shih HY, Chen JK, Lin CH, et al. Shared and restricted T‑cell receptor use is crucial for carbamazepine‑induced Stevens‑Johnson syndrome. J Allergy Clin Immunol 2011;128:1266‑76. e11 [ Links ]
27. Pichler WJ, Watkins S. Interaction of small molecules with specific immune receptors: The p‑I concept and its consequences. Curr Immunol Rev 2014;10:7-18. [ Links ]
28. Mallal S, Nolan D, Witt C, Masel G, Martin AM, Moore C, et al. Association between presence of HLA‑B* 5701, HLA‑DR7, and HLA‑DQ3 and hipersensitivity to HIV‑1 reverse‑transcriptase inhibitor abacavir. Lancet 2002;359:727‑32. [ Links ]
29. Hetherington S, Hughes AR, Mosteller M, Shortino D, Baker KL, Spreen W, et al. Genetic variations in HLA‑B region and hypersensitivity reactions to abacavir. Lancet 2002;359:1121‑2. [ Links ]
30. Mallal S, Phillips E, Carosi G, Molina J‑M, Workman C, Tomazic J, et al. HLA‑B* 5701 screening for hypersensitivity to abacavir. N Engl J Med 2008;358:568‑79. [ Links ]
31. Chessman D, Kostenko L, Lethborg T, Purcell AW, Williamson NA, Chen Z, et al. Human leukocyte antigen class I‑restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity 2008;28:822‑32. [ Links ]
32. Gonçalo M, Coutinho I, Teixeira V, Gameiro A, Brites M, Nunes R, et al. HLA‑B* 58:01 is a risk factor for allopurinol induced DRESS and SJS/TEN in a portuguese population. Br J Dermatol 2013; 169:660‑5. [ Links ]
33. Wei CY, Chung WH, Huang HW, Chen YT, Hung SI. Direct interaction between HLA‑B and carbamazepine activates T cells in patients with Stevens‑Johnson syndrome. J Allergy Clin Immunol 2012;129:1562–9. [ Links ]
34. Norcross MA, Luo S, Lu L, Boyne MT, Gomarteli M, Rennels AD, et al. Abacavir induces loading of novel self‑peptides into HLA‑B* 57: 01: an autoimmune model for HLA‑associated drug hypersensitivity. AIDS 2012;26:F21-9. [ Links ]
35. Adam J, Wuillemin N, Watkins S, Jamin H, Eriksson KK, Villiger P, et al. Abacavir induced T cell reactivity from drug naive individuals shares features of allo‑immune responses. PLoS One 2014;9:e95339 [ Links ]
36. Tohyama M, Hashimoto K, Yasukawa M, Kimura H, Horikawa T, Nakajima K, et al. Association of human herpesvirus 6 reactivation with the flaring and severity of drug‑induced hypersensitivity syndrome. Br J Dermatol 2007;157:934‑40. [ Links ]
37. Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Peer I, Floratos A, et al. HLA‑B* 5701 genotype is a major determinant of drug‑induced liver injury due to flucloxacillin. Nat Genet 2009;41:816‑9. [ Links ]
38. Wuillemin N, Adam J, Fontana S, Krähenbühl S, Pichler WJ, Yerly D, et al. HLA haplotype determines hapten or p‑I T cell reactivity to flucloxacillin. J Immunol 2013; 190: 4956‑64. [ Links ]
39. Matzinger P. Tolerance, danger and the extended family. Annu Rev Immunol 1994;12:991‑1045. [ Links ]
40. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti‑viral compounds activate immune cells via the TLR7 MyD88‑dependent signaling pathway. Nat Immunol 2002;3:196‑200. [ Links ]
41. Naisbitt DJ, Farrell J, Gordon SF, Maggs JL, Burkhart C, Pichler WJ, et al. Covalent binding of the nitroso metabolite of sulfamethoxazole leads to toxicity and major histocompatibility complex‑restricted antigen presentation. Mol Pharmacol 2002;62:628‑37. [ Links ]
42. Rodriguez‑Pena R, Lopez S, Mayorga C, Antunez C, Fernandez TD, Torres MJ, et al. Potential involvement of dendritic cells in delayed‑type hypersensitivity reactions to beta‑lactams. J Allergy Clin Immun 2006;118:949‑56. [ Links ]
43. Elsheikh A, Lavergne SN, Castrejon JL, Farrell J, Wang H, Sathish J, et al. Drug antigenicity, immunogenicity, and costimulatory signaling: Evidence for formation of a functional antigen through immune cell metabolism. J Immunol 2010;185:6448‑60 [ Links ]
44. Lavergne SN, Wang H, Callan HE, Park BK, Naisbitt DJ. Danger conditions increase sulfamethoxazole‑protein adduct formation in human antigen‑presenting cells. J Pharmacol Exp Ther. 2009; 331:372‑81. [ Links ]
45. Pleasants RA, Walker TR, Samuelson WM. Allergic reactions to parenteral beta‑lactam antibiotics in patients with cystic fibrosis. Chest 1994;106:1124‑8. [ Links ]
46. Milpied‑Homsi B, Moran EM, Phillips EJ. Antiviral drug allergy. Immunol Allergy Clin North Am 2015;34:645‑62. [ Links ]
47. Slatore CG, Tilles SA. Sulfonamide hypersensitivity. Immunol Allergy Clin North Am 2004;24:477‑90. [ Links ]
48. Shiohara T, Inaoka M, Kano Y. Drug‑induced hypersensitivity syndrome (DIHS): a reaction induced by a complex interplay among herpesviruses and antiviral and antidrug immune responses. Allergol Int 2006;55:1‑8. [ Links ]
49. Pavlos R, Mallal S, Ostrov D, Pompeu Y, Phillips E. Fever, rash, and systemic symptoms: Understanding the role of virus and HLA in severe cutaneous drug allergy. J Allergy Clin Immunol Pract 2014;2:21‑33. [ Links ]
50. Elsheikh A, Castrejon L, Lavergne SN, Whitaker P, Monshi M, Callan H, et al. Enhanced antigenicity leads to altered immunogenicity in sulfamethoxazole‑hypersensitive patients with cystic fibrosis. J Allergy Clin Immunol 2011;127:1543‑51.e3. [ Links ]
51. Coombs R, Gell P. Classification of allergic reactions responsible for drug hypersensitivity reactions. In: Clinical aspects of immunology Coombs RRA, Gells, PGH (Eds). 1968: 575‑96. [ Links ]
52. Caubet J, Pichler WJ, Eigenmann PA. Educational case series : Mechanisms of drug allergy. Pediatric Allergy and Immunology 2011;22:559‑67. [ Links ]
53. Pumphrey R. Anaphylaxis: Can we tell who is at risk of a fatal reaction? Curr Opin Allergy Clin Immunol 2004;4:285‑90. [ Links ]
54. Commins SP, James HR, Kelly LA, Pochan SL, Workman LJ, Perzanowski MS, et al. The relevance of tick bites to the production of IgE antibodies to the mammalian oligosaccharide galactose‑a‑1,3‑galactose. J Allergy Clin Immunol 2011;127:1286‑93. [ Links ]
55. Chung W, Hung S, Yang J, Su S, Huang S, Wei C, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens‑Johnson syndrome and toxic epidermal necrolysis. Nat Med 2008;14:1343‑50. [ Links ]
56. Pichler WJ, Adam J, Daubner B, Gentinetta T, Keller M, Yerly D. Drug hypersensitivity reactions: Pathomechanism and clinical symptoms. Medical Clin North Am 2010;94:645‑64. [ Links ]
57. Jerschow E, Lin RY, Scaperotti MM, Mcginn AP. Fatal anaphylaxis in the United States, 1999‑2010: Temporal patterns and demographic associations. J Allergy Clin Immunol 2014;134: 1318‑28.e7. [ Links ]
Frederico S. Regateiro
Serviço de Imunoalergologia
Centro Hospitalar Universitário de Coimbra
Praceta Prof. Mota Pinto
3000‑075 Coimbra, Portugal
Declaração de apoios financeiros: Nada a declarar.


{kind=link}
{kind=link}