










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.27 no.4 Lisboa dez. 2013
ORIGINAL ARTICLE
Disruption of urate transport in familial renal glucosuria and report on SGLT2 expression in normal and pathological kidney
Alteração do transporte de ácido úrico na Glicosúria Renal Familiar e expressão de SGLT2 no rim normal e patológico
Ines Aires1,2, Ana Rita Santos3, Roberto Bogarin4, Gurkan Genc5, Jorge Pratas6, Ozan Ozkaya5, Fernanda Carvalho3, Jose Rueff1, Fernando Nolasco2, Joaquim Calado1,2.
1 Department of Genetics Lisbon, Faculty of Medical Sciences, NOVA University of Lisbon. Lisbon, Portugal
2 Department of Nephrology, Hospital Curry Cabral. Lisbon, Portugal
3 Department of Renal Pathology, Hospital Curry Cabral. Lisbon, Portugal
4 Department of Pediatric Endocrinology, Hospital Nacional de Niños San José. Costa Rica
5 Department of Pediatric Nephrology, Ondokuz Mayis University Faculty of Medicine Samsun. Turkey
6 Department of Nephrology, Coimbra University Hospital. Coimbra, Portugal
ABSTRACT
Familial renal glucosuria (FRG) is a rare co-dominantly inherited benign phenotype characterized by the presence of glucose in the urine. It is caused by mutations in the SLC5A2 gene that encodes SGLT2, a Na+ -glucose co -transporter. The purpose of our current work was twofold: to characterize the molecular and phenotype findings of an FRG cohort and, in addition, to detail the SGLT2 expression in the adult human kidney. The phenotype of FRG pedigrees was evaluated using direct sequencing for the identification of sequence variations in the SLC5A2 gene. The expression of SGLT2 in the adult human kidney was studied by immunofluorescence on kidney biopsy specimens. In the absence of renal biopsies from FRG individuals, and in order to evaluate the potential disruption of SGLT2 expression in a glucosuric nephropathy, we have selected cases of nucleoside analogues induced proximal tubular toxicity. We identified six novel SLC5A2 mutations in six FRG pedigrees and described the occurrence of hyperuricosuria associated with hypouricaemia in the two probands with the most severe phenotypes. Histopathological studies proved that SGLT2 is localized to the brush -border of the proximal tubular epithelia cell and that this normal pattern was found to be disrupted in cases of nucleoside analogues induced tubulopathy. We present six novel SLC5A2 mutations, further contributing to the allelic heterogeneity in FRG, and identified hyperuricosuria and hypouricaemia as part of the FRG phenotype. SGLT2 is localized to the brush -border of the proximal tubule in the adult human normal kidney, and aberrant expression of the co-transporter may underlie the glucosuria seen with the use of nucleoside analogues.
Key words: glucosuria; proximal tubule; SGLT2 expression; uric acid.RESUMO
A Glicosúria renal familiar (GRF) é um fenótipo de transmissão autossómica co -dominante, causada por mutações do gene SLC5A2 que codifica SGLT2, o principal co -transportador de Na+ -glucose do túbulo proximal. Caracteriza-se pela redução da reabsorção tubula renal de glucose, causando glicosúria na ausência de hiperglicemia e de outros sinais de disfunção tubular. O objectivo do trabalho aqui apresentado foi caracterizar a nível molecular e fenotípico um conjunto de famílias afectadas por esta entidade e ainda detalhar a expressão de SGLT2 in vivo em amostras de tecido renal normal e patológico. Avaliámos fenotipicamente um grupo de indivíduos afectados por GRF e identificámos por sequenciação directa as alterações encontradas ao gene SLC5A2. A expressão de SGLT2 no rim adulto humano foi estudada por microscopia de fluorescência. Na ausência de biópsias renais de indivíduos afectados com GRF e de modo a avaliar potenciais alterações de expressão de SGLT2 em nefropatias com glicosúria, seleccionamos casos de lesão tubular proximal por toxidade de análogos de nucleósidos. Identificamos seis novas mutações de SLC5A2 em indivíduos de seis famílias. Nos indivíduos com glicosúria mais severa observamos igualmente hiperuricosúria com hipouricemia. Pudemos demonstrar que no rim adulto normal, SGLT2 está presente na bordadura em escova do epitélio do tubo proximal e que em casos de tubulopatia dos análogos dos nucleósidos a sua expressão está diminuída. Apresentamos assim seis novas mutações de SLC5A2 que contribuem para a heterogeneidade alélica da GRF e identificámos a hiperuricosúria com hipouricemia como características deste fenótipo. Confirmámos a expressão de SGLT2 na bordadura em escova do túbulo proximal e demonstramos que a diminuição da sua expressão pode estar subjacente à glicosúria observada na terapêutica com análogos dos nucleósidos.
Palavras chave: ácido úrico; expressão de SGLT2; glicosúria; tubo proximal.
INTRODUCTION
Familial renal glucosuria (FRG) is characterized by the presence of glucose in the urine in the absence of diabetes mellitus or generalized proximal tubular dysfunction. Mutations in the SLC5A2 gene, positioned in 16p11.2 and encoding SGLT2, the Na+-coupled glucose transporter that accounts for the bulk of glucose reabsorption in the renal proximal tubule, are responsible for the large majority of FRG pedigrees1. Most of these mutations are private and usually involve missense alleles, although nonsense, small deletions (both in-frame and frameshift) and splice site mutations have been reported. Even though an increasing number of FRG cohorts is being reported2-5, patients presenting with severe forms of FRG with glucosuria in excess of 50 g/1.73m2/24h are distinctly rare.
Two members of the Na+ -coupled glucose transporter family, SGLT2 and (to a lesser extend) SGLT1, are responsible for the secondary active uptake of glucose from the proximal lumen at the apical side of the epithelia. Once within the cell, glucose is returned to the systemic circulation by facilitative transport mediated by GLUT1 and 26.
Renal tubular glucose transport has been pointed out as a target in the treatment of type 2 diabetes mellitus (T2DM) and several SGLT2 inhibitors (SGLT2i) are now awaiting FDA approval. In a previous report, we anticipated that these compounds were likely to have a diuretic effect, based on the findings of moderate volume contraction with secondary activation of the renin -angiotensin-aldosterone system in severe FRG phenotypes2,3. This was later ascertained in clinical trials with SGLT2i7,8 .
Our group performs routine molecular characterization of FRG pedigrees and, whenever possible, phenotypic evaluation of such individuals. We now report the molecular and clinical findings of six pedigrees displaying novel SLC5A2 alleles. For two of these probands, glucosuria was in the range of the urinary glucose excretion (UGE) induced by pharmacological inhibition of SGLT2. One interesting and consistent observation with the administration of SGLT2i, is the almost dose-proportional decrease in serum uric acid levels7,8, which is paradoxical in light of the previously mentioned diuretic effect. We have, therefore, evaluated severe FRG phenotypes regarding serum and urinary uric acid levels. In addition, we assessed SGLT2 protein expression in adult normal human kidney and validated an FITC-immunofluorescence technique for paraffin -embedded kidney biopsy samples, with the purpose of detailing the topological location of SGLT2. Both techniques were performed using an available commercial antibody. In the absence of renal specimens from FRG individuals, we have searched for aberrant SGLT2 expression in renal biopsies of patients with nucleoside analogues induced proximal tubular toxicity and glucosuria.
SUBJECTS AND METHODS
Phenotype evaluation and mutation analysis of FRG pedigrees
This study was part of the medical evaluation for glucosuria. After obtaining informed consent from participating individuals or their legal guardians, individuals were enrolled. Mutation analysis was performed as previously reported9, but for exons 10 to 12, a single amplicon of 978 bp was PCR amplified and directly sequenced, using the following forward 5 - CTCGTCGTGAAGCTCATGC -3 and reverse 5 - CCAACCCTCAGTCGAGAAAT -3 primers. Only individuals bearing SLC5A2 mutations not previously described were included in this report.One hundred control chromosomes were assayed for the presence of the missense mutations identified, excluding them as common polymorphisms. We used direct sequencing of exons 5 and 12, for the c.571 A>C and c.1475 G>T alleles, respectively. In the case of the c.1343 A>T transversion, a new TspRI restriction site was inserted within the 978 bp exon 10 -12 amplicon, therefore enabling easy testing by PCR-RFLP of affected members and control chromosomes.
Immunohistology of kidney biopsies
SGLT2 detection by immunofluorescence was performed in five formalin -fixed and paraffin-embedded kidney biopsies with no detectable histopathological changes. Two additional sample of known proximal tubular toxicity with glucosuria induced by tenofovir or adefovir were also included. Heat -induced epitope retrieval was applied to 3μm sections with Target Retrieval Solution pH 9 (ref. S2367, Dako) at 95-99°C for 20 min. Incubations with primary goat polyclonal antibody anti -human SGLT2 C-terminal domain, from residues 590-640 (ref sc-47401, from SantaCruz Biotechnology (1:5) and a secondary antibody donkey anti-goat IgG - FITC (1:20) – ref sc-2420, Santa Cruz Biotechnology – were carried out for 60 ` at room temperature.
RESULTS
Phenotype evaluation and mutational analysis
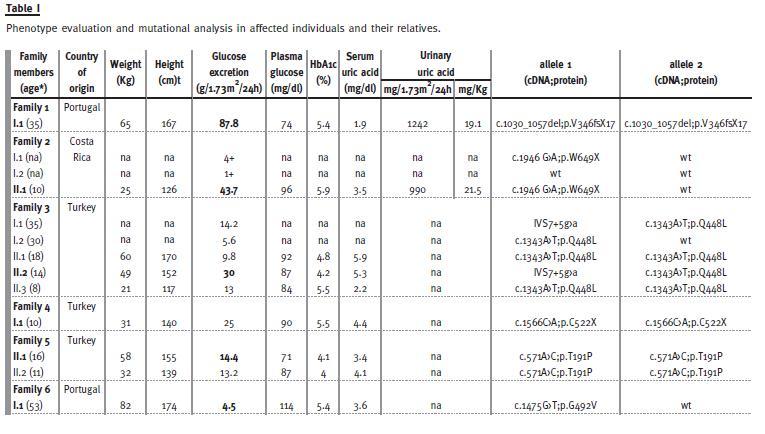
We identified six novel alleles in 13 individuals, from six different pedigrees. The available clinical data is detailed in Table I. Relatives were screened for glucosuria by dipstick urine analysis and, whenever possible, quantified by a 24h urine collection. All affected individuals had normal renal function.
Two heterozygous, one compound heterozygous and three homozygous probands were identified. The two probands with the most severe phenotypes, with UGE of 87.8 and 43.7 g/1.73m2/24h – individuals I.1 of pedigree 1 and II.1 of pedigree 2 – also had hyperuricosuria: excreting 1242 and 990 mg/1.73m2/24h (reference values 250 -750), with an urinary uric acid (mg)/Kg of body weight of 19.1 and 21.5 (reference values in a healthy population: 7.6}3.75 [adolescents] and 7}1.6 [adults]10, respectively. For proband of pedigree 1 (the most severe of our cohort) this was clearly associated with hypouricaemia of 1.9 mg/dl (reference range: 2.5 -7). Proband II.1 of family 2, although having a serum uric acid of 3.5 mg/dl at the time of evaluation, by the age of eight years the reported value was of 2.4 mg/dl.
Three mutated alleles are missense substitutions: p.T191P, p.Q448L and p.G492V. All represent non-conservative substitutions (with the exception of Val for Gly, in that both are non -polar aminoacids) affecting highly conserved residues among SGLT members (Fig. 1). Their putative pathogenicity was further assessed in silico using the bioinformatic algorithm Polyphenand, as such, all were predicted to be probably damaging. In addition, these sequence variations were not detected in 100 control chromosomes and were, therefore, excluded as common polymorphisms. There are three truncating mutations, including two non-sense, p.C522X and p.W649X and one frameshift, p.V346fsX17.
Family 3 displays a unique genetic feature. The proband, II.2, is a 14 - year -old girl born of a consanguineous marriage. Yet, her severe phenotype is not related to consanguinity but to compound heterozygosity: in addition to the ancestral p.Q448L mutation, shared by both parents, she displays the donor splice site mutation IVS7+5g>a transmitted by the father, in trans with the former allele. The remaining siblings are homozygous for the ancestral p.Q448L mutation, as anticipated in consanguineous marriages.
SGLT2 expression in human kidney
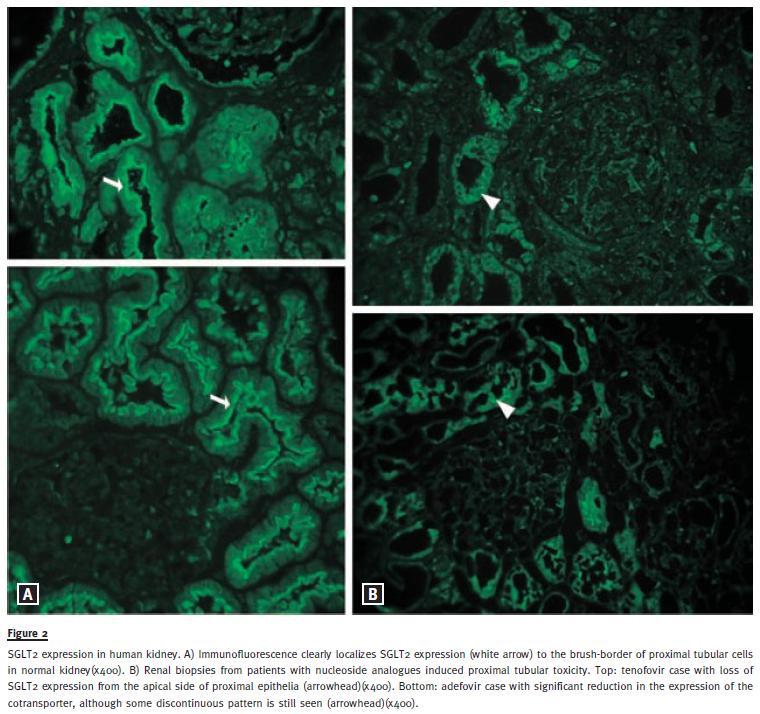
Immunofluorescence experiments in five kidney biopsies with no detectable histopathological changes localized SGLT2 to the brush -border of proximal tubular epithelia in normal kidney (Fig. 2A). Two patients with nucleoside analogues induced tubulopathy and glucosuria were identified in our pathological database. The first patient presented with acute renal failure after the introduction of tenofovir for his HIV condition. At renal biopsy, serum creatinine (Scr) was 7.0 mg/dl (on dialysis) and the 24h UGE of 4.9 g (175 mg/dl). The second individual had been for several months on adefovir for hepatitis B infection, before referral to our department. At evaluation, Scr was 1.7 mg/dl and the 24h UGE of 15.4 g (358 mg/dl). By immunofluorescence, apical SGLT2 expression was abolished in the biopsy of first patient, while in the second case it was significantly reduced (Fig. 2B).
DISCUSSION
The kidney contributes to glucose homeostasis by reabsorbing at the proximal tubule virtually all of the glucose filtered by the glomerulus, in a two – step process.
Firstly, glucose and Na+ are sequentially cotransported from the tubular lumen via secondary active transport mediated by the renal Na+ -glucose co-transporters SGLT2 and SGLT1. Secondly, glucose is released from tubular epithelial cells into the systemic circulation by facilitative Na+ -independent glucose transporters GLUT1 and GLUT2. SGLT2 is a low-affinity, high -capacity transporter, predominately expressed at the most proximal S1 tubular segment of the nephron11. Under physiological conditions, SGLT2 reabsorbs the majority of the filtered glucose12 and is considered a candidate gene for FRG. Mutations in SGLT1 account for the glucose -galactose malabsorption syndrome (GGM), which is characterized by severe diarrhoea upon the introduction of glucose or galactose to the diet of infants13. The different phenotypes of FRG and GGM reflect the differential expression pattern of both co-transporters as assessed by RT-PCR: SGLT1 being expressed mainly in intestinal epithelia, while SGLT2 is mostly restricted to the proximal renal tubule14. Another important difference between both cotransporters is the apparent difficulty in assessing SGLT2 expression by immunodetection, as compared to SGLT1. In fact, most of the work for SGLT2 was done by in -situ hybridization or RT-PCR11,14.
This report aims at further clarifying the renal tubular glucose transport by reporting the phenotype and genotype findings of FRG pedigrees displaying novel SGLT2 alleles, particularly for those individuals with severe forms, and also by assessing SGLT2 protein expression in adult human kidney.
For the six pedigrees analysed, there are six novel SLC5A2 alleles and the IVS7+5g>a mutation already reported. According to the previously described co -dominant pattern of inheritance for FRG, those individuals homozygous or compound heterozygous for a given mutation were shown to have larger UGE, when compared to heterozygous patients. For this cohort, the previously defined 10 g/1.73m2/24h cut-off appropriately seems to apply, with the exception of proband from pedigree 2. In this individual having an unusual severe phenotype, only one mutation was characterized. In fact, a second sequence variation in intron 3 (IVS3 -20g>a [data not shown]) was identified, but after testing the remaining family members, it was found to be in cis with the nonsense mutation. It is possible that the second allele might have been entirely missed, either because it resides outside the coding region or it consists on a large DNA rearrangement not amenable to detection by routine PCR based techniques. Moreover, a dominant negative effect or transheterozygosity with another gene coding for a protein also having a role in renal glucose transport, are alternative explanations.
All characterized sequence variations are expected to be deleterious for SGLT2 function: the three missense mutations are predicted to be pathogenic substitutions, while the p.V346fsX17, p.C522X and p.W649X mutations, expected to lead to truncated proteins at SGLT2 transmembrane domains 3, 13 and 14, respectively. The IVS7+5g>a allele of Family 3 is recurrently reported in unrelated pedigrees of different ethnic origins15. Considering the significant allelic heterogeneity in FRG, with most mutations being family specific, the high recurrence for this single allele can be due to a mutational hot-spot. This pedigree represents the first instance of allelic heterogeneity in a consanguineous FRG pedigree, in that the proband is a compound heterozygous for the ancestral p.Q448L mutation, shared by both parents, and the splice site mutation in intron 7. This family furthermore illustrates the co-dominant nature of FRG, with all members being affected irrespectively of heterozygosity (mother), compound heterozygosity (father and proband) or homozygosity (remaining siblings) for the identified mutations.
Hyperuricosuria with hypouricemia is the most interesting phenotype finding in the probands presenting with the highest UGE, suggesting urinary urate over -excretion as the mechanism underlying hypouricaemia in FRG and in farmacological SGLT2 inhibition7,8. These observations also rule out the possibility of non-specific pharmacological inhibition of renal urate transporters like GLUT9, the facilitative glucose transporter encoded by the SLC2A9 gene, mutated in familial renal hypouricemia16 and expressed at the proximal renal tubule in humans.
Interestingly, GLUT9 was also shown to exchange extracellular glucose for intracellular urate17. Two isoforms, GLUT9S (expressed at the apical side) and GLUT9L (at the baso -lateral side) act concertedly to reabsorb uric acid from the glomerular filtrate. However, in case of a high glucose concentration in the proximal tubular lumen, as seen in FRG and with SGLT2 pharmacological inhibition, the direction of urate transport could be reversed, with glucose being exchanged for urate and leading to excessive urinary excretion and secondary hypouricaemia.
We have detailed the expression of SGLT2 in human adult kidney by assessing its expression by immunofluorescence in biopsy specimens. A previous report4 had already localized SGLT2 to the apical side of proximal renal tubular epithelia in humans. Our experiments confirm that SGLT2 is, in fact, expressed at apical side of normal proximal tubular epithelia and confined to the brush-border. Importantly, its expression is distinctly reduced or absent from the apical side of the proximal tubule in the cases of antiretroviral renal toxicity, both presenting with proximal tubulopathy with glucosuria. These anticipated negative controls not only confirm the specificity of the antibody used, but also identify the loss for SGLT2 expression as the mechanism underlying the glucosuria induced by some nucleoside analogues.
In conclusion, we present the phenotype and genotype characterization of six FRG pedigrees displaying six novel SGLT2 mutations. The hyperuricosuria found in FRG is a plausible direct cause for the hypouricaemia in the setting of severe glucosuria, either occurring naturally -as in FRG - or induced by pharmacological inhibition of SGLT2. We theorized about GLUT9 being the transporter responsible for these observations, by means of an antiporter mechanism. Finally, we localized SGLT2 in the human adult normal kidney, to the brush -border of proximal tubular epithelia cell.
References
1. Santer R, Kinner M, Lassen CL, et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol 2003;14(11):2873 -2882. [ Links ]
2. Calado J, Loeffler J, Sakallioglu O, et al. Familial renal glucosuria: SLC5A2 mutation analysis and evidence of salt -wasting. Kidney Int 2006;69(5):852 -855. [ Links ]
3. Calado J, Sznajer Y, Metzger D, et al.Twenty -one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant 2008;23(12):3874-3879. [ Links ]
4. Yu L, Lv JC, Zhou XJ, Zhu L, Hou P, Zhang H. Abnormal expression and dysfunction of novel SGLT2 mutations identified in familial renal glucosuria patients. Hum Genet 2011;129(3):335-344. [ Links ]
5. Lee H, Han KH, Park HW, et al. Familial renal glucosuria: a clinicogenetic study of 23 additional cases. Pediatr Nephrol 2012;DOI:10.1007/s00467 -012 -2109-2109. [ Links ]
6. Mather A, Pollock C. Glucose handling by the kidney. Kidney Int 2011; 79(Suppl 120):S1-S6. [ Links ]
7. List JF, Woo V, Morales E, Tang W, Fiedorek FT.. Sodium -glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care 2009;32(4):650 -657. [ Links ]
8. Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type2 diabetes who have inadequate glycemic control with metformin: a randomized, double blind, placebo–controlled trial. Lancet 2010;375(9733):2223-2233. [ Links ]
9. Calado J, Soto K, Clemente C, Correia P, Rueff J. Novel compound heterozygous mutations in SLC5A2 are responsible for autosomal recessive renal glucosuria. Hum Genet 2004;114(3):314–316. [ Links ]
10. Baldree LA, Stapleton FB. Uric acid metabolism in children. Pediatr Clin North Am 1990;37(2):391-418. [ Links ]
11. Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D -glucose. J Clin Invest 1994;93(1):397-404. [ Links ]
12. Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 2011;22(1):104-112. [ Links ]
13. Martin MG, Turk E, Lostao MP Kerner C, Wright EM. Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose -galactose malabsorption. Nat Genet 1996;12(2):216-220. [ Links ]
14. Chen J, Williams S, Ho S, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther 2010;1(2):57-92. [ Links ]
15. Calado J, Santer R, Rueff J: Effect of kidney disease on glucose handling (including genetic defects). Kidney Int 2011; 120 (Suppl 7): S1–S6. [ Links ]
16. Matsuo H, Chiba T, Nagamori S, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet 2008;83: c744 -c781. [ Links ]
17. Caulfield MJ, Munroe PB, ONeill D, et al. SLC2A9 is a high -capacity urate transporter in humans. PLoS Med 2008;5(10):e197. [ Links ]
Dra Ines Aires
Department of Genetics Lisbon,
NOVA University of Lisbon, Portugal
Rua da Junqueira, 100
1349–008, Lisboa, Portugal
E-mail: inesaires@yahoo.com
Acknowledgments
We thank all family members for their participation. We are grateful to Jorge Gaspar for providing the DNAs of control chromosomes. This work was supported and partially funded by APENE – Associacao Portuguesa para o Estudo das Nefropatias.
Conflict of interest statement: None declared.
Received for publication: 30/07/2013
Accepted in revised form: 24/10/2013


{kind=link}
{kind=link}
{kind=link}