










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.28 no.1 Lisboa mar. 2014
CASE REPORT
Primary hyperparathyroidism in young patients – a challenging cause of hypercalcaemia
Hiperparatiroidismo primário em doentes jovens – uma causa desafiante de hipercalcemia
Pedro Azevedo1, Cristina Freitas1, Susana Garrido2, Cláudia Amaral2, João Pedro Pimentel1, Rui Carvalho2, António Cabrita1
1 Department of Nephrology, Centro Hospitalar do Porto – Santo António Hospital. Porto, Portugal.
2 Department of Endocrinology, Centro Hospitalar do Porto – Santo António Hospital. Porto, Portugal.
ABSTRACT
Hypercalcaemia is rare in the clinical practice and is often a clue to the presence of an unsuspected illness.
Hyperparathyroidism and malignancy account for 80 -90% of hypercalcaemia states. Primary hyperparathyroidism (PHPT) results from an autonomous overproduction of parathyroid hormone (PTH) and it occurs most commonly in over 50-year-old individuals and in post -menopausal women. On rare occasions, it may be a feature of a familial condition, such as multiple endocrine neoplasia type 1 syndrome. The diagnosis of PHPT is frequently delayed because it is commonly asymptomatic and is often identified only after development of severe signs and symptoms, such as bone fractures, with significant morbidity. Parathyroidectomy is the treatment of choice, as it is effective at restoring normal serum calcium and PTH levels and has few complications. We report the case of a young patient presenting with severe hypercalcaemia, a solitary brown tumour and pathological bone fracture that led to the diagnosis of PHPT. A brief review of the current literature on this topic is performed.
Key-words: Hypercalcaemia; hyperparathyroidism; hungry bone syndrome.
RESUMO
A hipercalcemia é rara na prática clínica e é, muitas vezes, sugestiva da presença de uma doença não suspeita. O hiperparatiroidismo e as neoplasias são responsáveis por 80 -90% dos casos de hipercalcemia.
O hiperparatiroidismo primário (HPTP) resulta da hiperprodução autónoma da hormona paratiroideia (PTH) e ocorre habitualmente em doentes com mais de 50 anos e mulheres na pós -menopausa. Raramente, o HPTP pode ser uma manifestação de uma síndrome familiar, como a síndrome de neoplasia endócrina múltipla tipo 1. O diagnóstico do HPTP é, muitas vezes, tardio uma vez que se trata de uma doença frequentemente assintomática, sendo apenas identificada após aparecimento de sinais e sintomas graves, como fracturas ósseas, que condicionam morbilidade significativa. A paratiroidectomia constitui o tratamento de escolha, uma vez que é eficaz na normalização dos níveis de cálcio e PTH e tem poucas complicações.
Os autores descrevem o caso de um doente jovem com hipercalcemia grave, com tumor castanho e uma fratura patológica que conduziram ao diagnóstico de HPTP. Partindo do caso clínico, realizou -se uma breve revisão da literatura.
Palavras chave: Hipercalcemia; hiperparatiroidismo; síndrome osso faminto.
INTRODUCTION
Hypercalcaemia is a rare finding in the clinical practice, affecting between 0.1-1% of the general population and it is often a clue to the presence of an unsuspected illness. Hyperparathyroidism (HPT) and malignancy account for 80-90% of hypercalcaemic states1.
Other causes of hypercalcaemia, including vitamin D intoxication, sarcoidosis, tuberculosis, some fungal infections, thyrotoxicosis, Addisons disease, milk-alkali syndrome, vitamin A intoxication, therapy with thiazide diuretics or lithium carbonate, familial hypocalciuric hypercalcaemia, prolonged immobilization in patients with high skeletal turnover, and the recovery phase of rhabdomyolysis-associated acute renal failure, all amount to fewer than about 10% of all causes of hypercalcaemia2.
In ambulatory settings, HPT accounts for 50-60% of hypercalcaemia and for 27% or less in the hospital1.
By contrast, malignancy accounts for 31% or less of ambulatory hypercalcaemia but for up to 65% of that seen in the hospital1. Carcinomas of the bronchus, breast, head and neck, urogenital tract, and multiple myeloma account for 75% of the hypercalcaemia in malignancy1.
Hyperparathyroidism diagnosis is made by finding an elevated PTH concentration or one that is within the normal range but inappropriately elevated, given the patients calcaemia. Some patients may even present with plasma calcium concentration within the normal range3. Hyperparathyroidism may be primary, resulting from an autonomous overproduction of parathyroid hormone which is not downregulated by the calcium-sensing receptor3, or secondary in patients with chronic kidney disease, where parathyroid hyperplasia relates to chronic kidney disease-mineral and bone disorder4. In other patients with advanced renal disease, hypercalcaemia may be due to progression from appropriate parathyroid hyperplasia to autonomous overproduction of PTH, a disorder called tertiary HPT4.
Primary hyperparathyroidism (PHPT) is rare in children and young adults, with a reported incidence of 2 -5 per 100.000 and without an apparent sex predilection5. On rare occasions, PHPT may be a feature of a familial condition, such as multiple endocrine neoplasia (MEN) type 1, MEN2A, or the HPT-jaw tumour syndrome, familial isolated hyperparathyroidism and familial hypocalciuric hypercalcaemia/neonatal severe HPT6.
Indeed, PHPT has been less studied in children and young adults, with only small case series and case reports in the literature7. Some studies showed a higher prevalence of multiglandular disease and more severe disease in younger patients, compared to adults8,9, while others reported no difference in clinical presentations7,10,11.
Overall, controversy continues regarding the role of and reliance on various technologies, such as pre -operative localization imaging, intraoperative parathyroid hormone level measurements, and minimally invasive surgery. Regarding the surgical treatment and prognosis, several authors report similar outcomes between younger and older patients, considering a similar disease entity in both age groups10,11.
In all age groups, PHPT bone disease is rarely overt. The incidence of brown tumours in patients with PHPT is 1.5 -1.7% and can affect any part of the skeleton. Presentation of PHPT with a pathological fracture is very uncommon4.
The authors report an unusual case of severe PHPT in a young patient presenting with a pathological fracture.
CASE REPORT
A 34-year-old man had no familiar or pathological history until 2008, when he experienced an episode of non -lithiasic acute pancreatitis, (with no access to analytical results, including ionized calcium). One year after, he developed progressive bone and joint pain and proximal muscle weakness that persisted for the following 3 years. He was treated with paracetamol, tramadol and non -steroidal anti -inflammatory drugs.
In July 2012, he was referred to Orthopaedics for intense pain in the right leg. Computed tomography (CT) showed an osteolytic lesion with a length of approximately 8 cm, in the middle third of the tibia, with a medullary soft tissue mass and cortical destruction.
A biopsy showed fibrous tissue with some giant cells, favouring the diagnosis of a reactive lesion, probably inflammatory, with no signs of malignancy.
One month later, the patient presented with a pathological right subtrochanteric fracture, and was submitted to surgical treatment. Analytical studyrevealed serum creatinine 1.98 mg/dL, (NR: 0.7 -1.2), serum urea 23 mg/dL (NR: 10 -50), hypercalcaemia, with serum ionized calcium 2.44 mmol/L (NR:1.13 -1.32) and serum total calcium 4.39 mmol/L (NR: 2.09 -2.42), hypophosphatemia with serum phosphorus 0.65 mmol/L (NR: 0.87 -1.45) and hyperparathyroidism, with PTH 3005 pg/mL (NR:15 -65). Arterial blood gas analysis revealed a metabolic acidosis (pH 7.2), with normal anion gap and serum bicarbonate 18.7 mmol/L (NR: 22 -26). Renal ultrasound showed kidneys with reduced parenchymo-sinusal differentiation, with no renal calculi. There was no clinical or analytical evidence of other causes of hypercalcaemia.
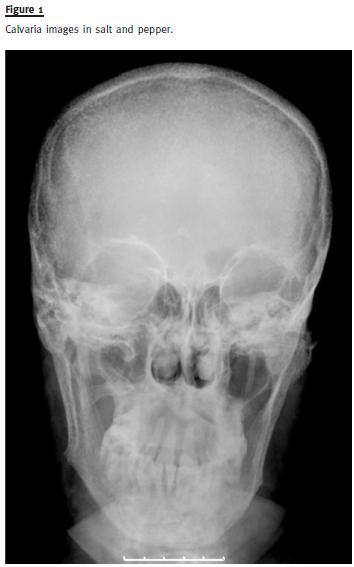
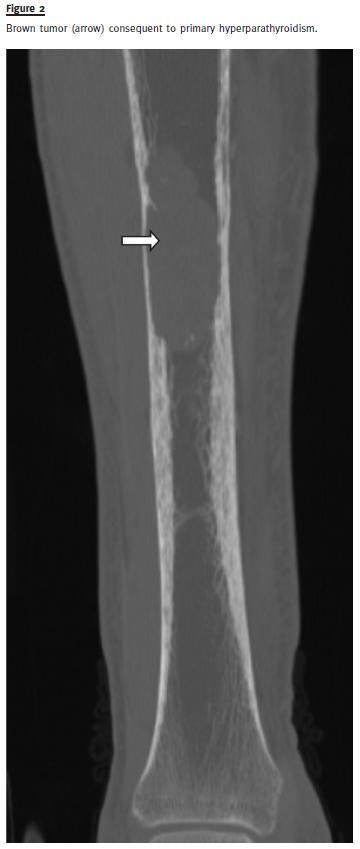
A skull X-ray revealed osteolytic lesions in typical salt and pepper pattern (Fig.1). The CT showed infiltrative bone disease with mixed lytic and sclerotic radiological pattern, suggestive of osteitis fibrosa cystica in the right tibia (Fig.2). In the topography of the parathyroids it was identified an expansive lesion (26x21mm) with a central necrotic area. Sestamibi scintigraphy confirmed a focus of hyperfixationin the left inferior parathyroid consistent with adenoma.
A left inferior parathyroidectomy and osteosynthesis of lytic lesion of the tibia were performed.
The histopathological result showed a parathyroid tumour of uncertain malignant potential. Parafibromin staining was not assessed.
Post-operatively, there was a progressive decline in the value of serum PTH (57pg/mL), serum calcium (ionized calcium of 0.8mmol/L), phosphorus (0.44mmol/L), magnesium (0.41mmol/L NR: 0.6 -1.1) and bicarbonate (11.3mmol/L, pH 7.2), consistent with the diagnosis of hungry bone syndrome.
The patient was treated with intravenous and oral supplementation of vitamin D, calcium, phosphorus, magnesium and bicarbonate. He improved, with normalization of ionic disturbances and progressive decline of serum creatinine levels to 1.4 mg/dL.
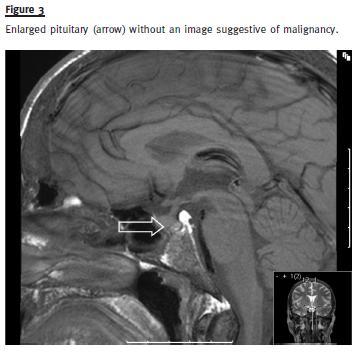
During hospitalization, the patient mentioned a diminished libido and impotence for the previous three years. Anterior pituitary hormone levels determination was compatible with hypogonadotropic hypogonadism: prolactin 30 ng/mL (NR:4 -15), FSH 0.7mUI/mL (NR:1.5 -12.4), LH 5.4 mUI/mL (NR:1.7 -8.6), testosterone 1.3 ng/mL (2.8 -8.0), TSH 2.8 uUI/mL (NR 0.2 -4.2), cortisol 21 ug/dL (NR 6.2 -19.4). Pituitary imaging revealed an enlarged pituitary with 13 mm (Fig.3). However, three months later and with no specific therapy, prolactin and testosterone levels returned to normal. There was no evidence of other endocrine tumours. Genetic testing for MEN1 mutation was negative.
The patient is being followed in Outpatient Nephrology and Endocrinology care. One year later, he is asymptomatic under calcium supplements, with stable serum creatinine of 1.4 mg/dl, normal calcium and phosphorus levels and PTH of 154 pg/mL. Updated bone scintigraphy is negative and the patient has normal testosterone, LH and FSH levels. These results suggest that the initial acute illness, probably, blunted the anterior pituitary hormone levels, returning to normal, with resolution of hyperparathyroidism and acute renal failure.
DISCUSSION
Primary hyperparathyroidism, with a prevalence of 0.8%, peaks in the seventh decade and occurs most commonly in post -menopausal women, but the incidence is similar in men and women before 45 years of age12. Head and neck irradiation in childhood and long-term lithium therapy have been associated with a greater prevalence of PHPT. The natural history of PHPT varies according to its severity, with worse disease in symptomatic patients who do not undergo surgery. Excepting rickets, which is found predominantly in younger patients8, the remaining clinical presentations and biochemical values are extremely variable between group ages8,11, with authors pointing to contradictory frequencies of serum calcium levels, kidney stones and musculoskeletal manifestations7,8,10.
Long -standing asymptomatic hypercalcaemia is the most common clinical presentation, with 75-80% of cases of PHPT diagnosed when a routine assay shows hypercalcaemia in patients who are asymptomatic or during evaluation for osteoporosis. In the other cases symptoms of hypercalcaemia are the clue to the diagnosis3.
Hypercalcemia may be associated with a spectrum of clinical manifestations, ranging from a few (if any) symptoms if the hypercalcaemia is mild and/or chronic to obtundation and coma if it is severe and/or acute. Clinical manifestations are neurological (altered mental status including lethargy, depression, confusion, stupor and coma), renal (impaired concentrating ability, dehydration, hypercalciuria, nephrolithiasis, nephrocalcinosis, distal renal tubular acidosis, acute and chronic renal insufficiency), musculoskeletal (muscle weakness, bone pain, increased bone resorption and fracture risk), cardiovascular (hypertension, shortened QT interval) and gastrointestinal (anorexia, constipation, nausea and vomiting)3.
Usually, when the patient shows signs or symptoms of hypercalcaemia, such as renal colic, nausea or vomiting, or even typical radiological abnormalities, they are treated individually, delaying the diagnosis of PHPT3. Furthermore, the sex and age of presentation of this patient may have downplayed the diagnosis.
Indeed, our patient presented, three years before, an episode of acute pancreatitis and prolonged bone pain, but it was a pathological fracture that led to further investigation. Although we were not able to confirm, the acute pancreatitis episode might also have been triggered by hypercalcaemia associated with a previous unidentified PHPT. The relationship between HPT and pancreatic inflammatory disease remains controversial, but it may be related to the deposition of calcium in the pancreatic duct and translation within the pancreatic parenchyma of inactive tripsinogen to trypsin by hypercalcaemia. Erdheim reported the first documented case of pancreatitis associated with HPT in 1903 in a post-mortem study [cit. by 13]. Hypocalcaemia is expected during an attack of pancreatitis and hypercalcaemia might be a clue for diagnosis of HPT. The treatment of HPT is essential in such cases to prevent relapses of pancreatitis.
Primary hyperparathyroidism has undergone a dramatic change in the latest 30 years, being predominantly an asymptomatic disorder. The development of classical findings, such as nephrolithiasis, brown tumours, bone cysts and pathological fractures is exceptional nowadays. Osteitis fibrosa cystica was initially described by Von Recklinghausen in 1891, but the aetiological link between the disease and parathyroid neoplasms was not established until 1925 [cit. by 13]. Typical radiological findings in PHPT patients include images of diffuse osteolytic lesions in typical salt and pepper mottling of skull, loss of lamina dura in the mandible, infiltrative bone disease with subperiostal bone resorption and lytic lesions with tortuous contours, known as brown tumours. Brown tumour is an extremely rare non-neoplastic osseous lesion (reported prevalence 0.1%) that constitutes a focal manifestation of osteitis fibrosa cystica induced by HPT, independently of its cause3. Because HPT is treated before such lesions develop, they are now uncommon. Brown tumour is mainly found in secondary HPT in patients with renal insufficiency on haemodialysis, but it has also been described as an extremely rare manifestation of PHPT.
Unlike our patient, it is more common in patients older than 50 years and three times more common in women. It may appear in any of the bones, but the long bones and jaw are the most common sites of these lesions. Clinically, brown tumours most often manifest as slowly growing, painful masses. These tumours can occasionally behave aggressively, multiple and destructive, mimicking metastases. It is imperative the distinction of PHPT with a neoplastic condition. In the latter, a low or undetectable PTH level rules out PHPT and raises the possibility of cancer-associated hypercalcaemia. If the PTH level is elevated in a person with a known malignant condition, the most likely diagnosis is concomitant HPT, as ectopic production of PTH -related protein (PTHrP) from a tumour is extremely rare.
As in our patient, the presence of PHPT and neuroendocrine symptoms in a young patient should lead to the search for other manifestations of a familial condition.
The MEN syndromes include varying combinations of more than 20 endocrine and non –endocrine tumours14 -18: MEN1 is a rare autosomal dominant disorder (incidence 1:30.000) with high penetrance, equal sex distribution and no ethnic group or racial predilection15-17. It is defined, clinically, by the occurrence of two or more primary tumours of the parathyroid glands (present in 95% of cases), anterior pituitary (15 -90% of cases) and endocrine gastroenteropancreatic tract (30 -80% of cases)14-16. Primary hyperparathyroidism linked to MEN1 represents 2–4% of all forms of PHPT19. MEN2A is characterized by the association of, at least, two of the classical clinical features: medullary thyroid cancer, pheochromocytoma and primary parathyroid hyperplasia. The initial suspicion of two endocrine tumours was not confirmed, with analytical and functional neuroendocrine normalization, concomitant with renal function recovery. Despite the initial suspicion of familial PTPH, our patients case of PHPT is probably sporadic, as investigation of the patient and first degree relatives did not conclude for any familial syndrome.
Parathyroid tumours have a malignant potential. The assessment of parafibromin staining in this patient would have been extremely useful, as parafibromin -positive atypical adenomas do not recur and can be considered benign. On the other hand, parafibromin-negative atypical adenomas should be considered tumours of low malignant potential, requiring a closer surveillance20.
The treatment of PHPT is primarily surgical. Operative intervention can be delayed in patients over 50 years of age who are asymptomatic or minimally symptomatic and who have serum calcium concentrations <1.0 mg/dL(0.2 mmol/L) above the upper limit of normal, and in patients who are medically unfit for surgery21.
If surgery is not recommended or the patient refuses it, then it is appropriate to recommend supportive-preventive measures and drug therapy, such as oestrogen-progestin therapy, bisphosphonates and calcimimetics. Other preventive measures include avoiding factors that can aggravate hypercalcaemia, physical activity to minimize bone resorption, adequate hydration, maintaining a moderate calcium intake (1000 mg/day) and moderate vitamin D intake (400 to 600 International Units daily)22.
Oestrogen-progestin therapy is beneficial in post-menopausal women with primary hyperparathyroidism because of its ability to reduce bone resorption. However, long -term oestrogen use is no longer recommended, given the associated cardiovascular risks and breast cancer23. Bisphosphonates may be useful in the long-term control of osteopenia in patients with untreated PHPT. Studies with alendronate revealed an increase in bone mineral density (BMD) at the hip and lumbar spine in patients with mild PHPT, but the increase in BMD was inferior to those seen after parathyroidectomy and levels of serum calcium and PTH did not change23. Cinacalcet, the only calcimimetic agent available for PHPT treatment, activates the calcium -sensing receptor in the parathyroid gland, thereby inhibiting PTH secretion. Studies revealed that, in patients with PHPT, cinacalcet reduced serum calcium (73 % of patients) and PTH concentrations (74-92% of patients)24. Furthermore, cinacalcet is approved for the treatment of moderate-to-severe hypercalcaemia in patients with PHPT unable to undergo parathyroidectomy.
However, cinacalcet it is not the medical equivalent of parathyroidectomy, because it does not provide symptom relief of PHPT, its effects are not permanent, PTH concentrations do not normalize and prolonged use may be associated with continued bone loss25. Furthermore, there are no data on the long-term effects on nephrolithiasis, neuropsychiatric, cognitive abnormalities and patients quality of life24.
Overall, parathyroidectomy is the treatment of choice and the only definitive therapy, achieving biochemical homeostasis and symptom relief10,11.
Moreover, it decreases the risk of nephrolithiasis, increases bone mineral density, and may decrease fracture risk and modestly improve some quality of life measurements.
Clinical surveillance and medical therapy in asymptomatic patients can be considered, and includes appropriate hydration, avoidance of diuretics, replacement of vitamin D deficiency and regular assessment of serum calcium levels in cases of prolonged immobilization26.
However, the current Guidelines recommend that surgery is indicated in asymptomatic patients who are younger than 50 years, have serum calcium concentration of 1.0 mg/dL or 0.25 mmol/L above the upper limit of normal, a 24 hour urinary calcium excretion greater than 400 mg or 10 mmol/day, creatinine clearance that is 30% lower than that of age-matched normal individuals, bone density at the hip, lumbar spine, or distal radius that is more than 2.5 standard deviations below peak bone mass (T score < -2.5) and/or previous fragility fracture or in patients in whom surveillance is not feasible21.
The traditional surgical procedure is bilateral neck exploration, which relies on visual and weight-based estimations of gland size to distinguish a single adenoma from multiglandular disease.
Although some authors report that 99mTc–sestamibi scintigraphy is less sensitive in younger patients, overall, it is the radionuclide study of choice for pre-operative evaluation, because of superior image quality, more favourable dosimetry and improved accuracy, localizing ectopic parathyroid glands [9]. Moreover, it can visualize up to 95% of parathyroid adenomas, allowing the use of less invasive procedures. Indeed, with increased experience, improved imaging modalities, and the use of adjuncts, such as intraoperative PTH monitoring27, minimally invasive parathyroidectomy (of only documented abnormal parathyroid gland) is emerging as the procedure of choice21. The cure rates of both approaches are similar in most paediatric and adult patients (95 -100%)28, but minimally invasive parathyroidectomy may be preferred, because of reduced operative times and less complications, such as injury to the recurrent laryngeal nerve and hypoparathyroidism [29]. Bilateral neck exploration is reserved in patients with a large goiter or previous neck surgery.
Recurrence is rare in sporadic PHPT14, but patients with MEN1 will usually require more than one surgery, when initial parathyroidectomy is conservative, and patients with familial hypocalciuric hypercalcaemia generally require no specific treatment30.
Hypocalcaemia is a common problem after parathyroidectomy.
The fall in serum calcium is primarily due to functional or relative hypoparathyroidism, leading to reduction in bone resorption, increase in bone formation with an increased influx of calcium into bone. In patients without end-stage renal disease, hypocalcaemia is due to increased calcium excretion and decreased calcium intestinal absorption due to reduced PTH -mediated renal 1,25(OH)2D production31.
The hypocalcaemia is generally transient because parathyroid tissue recovers function quickly. In some cases, however, the post-operative hypocalcaemia is severe and prolonged, despite normal or even elevated levels of parathyroid hormone. This phenomenon is called the hungry bone syndrome (HBS)31. The clinical features of the syndrome are largely due to hypocalcaemia and, to a lesser degree, hypophosphatemia, hypomagnesemia, and hyperkalemia. The serum calcium, typically, reaches a nadir two to four days post-operatively and may last up to three months31. Symptoms of hypocalcaemia include tetany, seizures, cardiac arrhythmias and laryngeal spasm. Careful monitoring of serum calcium is fundamental to prevent and treat symptomatic life-threatening hypocalcaemia. The hypomagnesemia can contribute to the development of refractory hypocalcaemia, by diminishing PTH secretion and inducing PTH resistance31. HBS should be anticipated pre-operatively and appropriate management instituted. Risk factors for the development of HBS still remain controversial. Older patients, higher pre-operative levels of serum calcium, increased levels of PTH and alkaline phosphatase have been associated with a higher risk of HBS32. These studies suggested that vitamin D deficiency and poorer nutritional intake in older patients promote a higher risk for the development of HBS32. However, a recent study showed that younger patients and patients with low pre-operative calcium levels were at higher risk for the development of HBS, hypothesizing that the increases in bone formation and osteoblasts after parathyroidectomy are more pronounced in younger versus older patients33.
There are no clear guidelines for the management of the HBS, but treatment is aimed at correcting each of the abnormalities (hypocalcaemia, hypomagnesemia, hypophosphatemia) with oral or intravenous supplementation of calcium, active metabolites or analogues of vitamin D and replenishment of magnesium stores, in order to restore a normal bone turnover31-33.
Our patient was last reviewed in the Nephrology Department two months ago. There were no new osteogenic symptoms and there has been sustained improvement in his musculoskeletal and sexual functions.
CONCLUSION
The authors report the case of a young male patient with advanced PHPT, diagnosed after a pathological bone fracture. The diagnosis of PHPT requires a high index of suspicion and the treatment involves medical-surgical measures. Parathyroidectomy performed after adequate localization is the treatment of choice with few complications. In the post -operative period, prevention and treatment of post-operative HBS are essential.
As PHPT is associated with significant morbidity, a multidisciplinary team approach, with endocrinological, nephrological, physical therapy and orthopaedic support is desirable for long-term optimal rehabilitation.
Longitudinal data are needed to better understand bone removal, fractures consolidation after parathyroidectomy and to assess the safety of the prolonged use of cinacalcet.
References
1. Lafferty FW. Differential diagnosis of hypercalcemia. J Bone Miner Res 1991;6 (Suppl 2):S51 -59. [ Links ]
2. Jacobs TP, Bilezikian JP. Clinical review: Rare causes of hypercalcemia. J Clin Endocrinol Metab 2005;90(11):6316–6322. [ Links ]
3. Gasser RW. Clinical aspects of primary hyperparathyroidism: clinical manifestations, diagnosis, and therapy http://www.ncbi.nlm.nih.gov/pubmed 2013;163(17-18):397-402. [ Links ]
4. Michels TC, Kelly KM. Parathyroid disorders.Am Fam Physician 2013:88(4):249-257. [ Links ]
5. Roizen J, Levine MA. Primary hyperparathyroidism in children and adolescents.J Chin Med Assoc 2012;75(9):425–434. [ Links ]
6. Hendy GN, Cole DE. Genetic defects associated with familial and sporadic hyperparathyroidism. Front Horm Res. 2013;41:149 -165. [ Links ]
7. Paunovic I, Zivaljevic V, Stojanic R, Kalezic N, Kazic M, Diklic A. Primary hyperparathyroidism in children and young adults: a single institution experience. Acta Chir Belg 2013;113(1):35-39. [ Links ]
8. Shah VN, Bhadada SK, Bhansali A, Behera A, Mittal BR, Bhavin V. Influence of age and gender on presentation of symptomatic primary hyperparathyroidism. J Postgrad Med 2012;58(2):107 -111. [ Links ]
9. Pashtan I, Grogan RH, Kaplan SP, et al. Primary hyperparathyroidism in adolescents: the same but different. Pediatr Surg Int. 2013;29(3):275 -279. [ Links ]
10. Joshua B, Feinmesser R, Ulanovski D, et al. Primary hyperparathyroidism in young adults. Otolaryngol Head Neck Surg 2004;131(5):628-632. [ Links ]
11. Sneider MS, Solorzano CC, Montano RE, Anello C, Irvin GL 3rd, Lew JI. Sporadic primary hyperparathyroidism in young individuals: different disease and treatment? J Surg Res 2009;155(1):100 -103. [ Links ]
12. Eufrazino C, Veras A, Bandeira F. Epidemiology of primary hyperparathyroidism and its non-classical manifestations in the city of Recife, Brazil. Clin Med Insights Endocrinol Diabetes 2013;6:69 -74. [ Links ]
13. Kelly TR. Relationship of hyperparathyroidism to pancreatitis. Arch Surg 1968;97(2):267-274. [ Links ]
14. Lairmore TC, Piersall LD, DeBenedetti MK, et al. Clinical genetic testing and early surgical intervention in patients with multiple endocrine neoplasia type 1 (MEN 1). Ann Surg 2004;239(5):637–645. [ Links ]
15. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97(9):2990 -3011. [ Links ]
16. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 2013 Aug 8. pii: S0303 -7207(13)00330-4. doi: 10.1016/j.mce.2013.08.002. [Epub ahead of print]. [ Links ]
17. Thakker RV. Multiple endocrine neoplasia type 1. Indian J Endocrinol Metab 2012;16(Suppl 2):272 -274. [ Links ]
18. Lu YY, Zhu F, Jing DD, et al. Multiple endocrine neoplasia type 1 with upper gastrointestinal hemorrhage and perforation: a case report and review. World J Gastroenterol 2013;19(8):1322 -1326. [ Links ]
19. Yamazaki M, Suzuki S, Kosugi S, et al. Delay in the diagnosis of multiple endocrine neoplasia type 1: typical symptoms are frequently overlooked. Endocr J 2012;59(9):797 -807. [ Links ]
20. Kruijjf S, Sidhu SB, Sywak MS, Gill AJ, Delbridge LW. Negative parafibromin staining predicts malignant behavior in atypical parathyroid adenomas. Ann Surg Oncol 2013 Oct1. [Epub ahead of print]. [ Links ]
21. Bilezikian JP, Khan AA, Potts JT Jr. Third International Workshop on the Managementof Asymptomatic Primary Hyperthyroidism. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 2009;94(2):335 -339. [ Links ]
22. Khan A, Grey A, Shoback D. Medical management of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 2009;94(2):373-381. [ Links ]
23. Parker CR, Blackwell PJ, Fairbairn KJ, Hosking DJ. Alendronate in the treatment of primary hyperparathyroid -related osteoporosis: a 2 -year study. J Clin Endocrinol Metab 2002;87(10):4482 -4489. [ Links ]
24. Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five -year study. J Clin Endocrinol Metab 2009;94(12):4860 -67. [ Links ]
25. Norman J, Lopez J, Politz D. Cinacalcet (Sensipar) provides no measurable clinical benefits for patients with primary hyperparathyroidism and may accelerate bone loss with prolonged use. Ann Sur Oncol. 2012 May; 19(5):1466 -71. [ Links ]
26. Calzada -Nocaudie M, Chanson B, Conte -Devolx B, et al. Prise en charge de lhyperparathyröidieprimaireasymptomatique. Ann. Endocrinol. 2006; 67(1):7-12. [ Links ]
27. Tonelli F, Marcucci T, Fratini G, et al. Is total parathyroidectomy the treatment of choice for hyperparathyroidism in multiple endocrine neoplasia type 1? Ann Surg. 2007; 246(6): 1075-1082. [ Links ]
28. Udelsman R, Pasieka JL, Sturgeon C, et al. Surgery for asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 2009; 94: 366. [ Links ]
29. Versnick M, Popadich A, Sidhu S, et al. Minimally invasive parathyroidectomy provides a conservative surgical option for multiple endocrine neoplasia type 1 - primary hyperparathyroidism. Surgery 2013;154(1): 101 -5. [ Links ]
30. Roizen J, Levine M. Primary hyperparathyroidism in children and adolescents. J Chin Med Assoc. 2012 September; 75(9): 425–434. [ Links ]
31. Brasier AR, Nussbaum SR. Hungry bone syndrome: clinical and biochemical predictors of its occurrence after parathyroid surgery. Am J Med 1988; 84: 654. [ Links ]
32. Witteveen JE, van Thiel S, Romijn JA, et al. Hungry bone syndrome: still a challenge in the post-operative management of primary hyperparathyroidism: a systematic review of the literature. Eur J Endocrinol. 2013 Feb 20;168(3):R45 -53. [ Links ]
33. Latus J, Roesel M, Fritz P, et al. Incidence of and risk factors for hungry bone syndrome in 84 patients with secondary hyperparathyroidism. Int J Nephrol Renovasc Dis. 2013 Jul 8;6:131 -137. [ Links ]
Dr. Pedro Azevedo
Rua D. António Barroso, nº 200 – Entrada 4 – 2 H
4050 -059 Porto
Portugal
E-mail: pedronunesazevedo@gmail.com
Conflict of interest statement: None declared.
Received for publication: 02/11/2013
Accepted in revised form: 1/01/2014

