










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.29 no.1 Lisboa mar. 2015
REVIEW ARTICLE
Focus on:
I – Hydro electrolyte balance Homeostasis
Pedro A. Correia
Department of Nephrology, Hospital Fernando da Fonseca. Amadora, Portugal
INTRODUCTION – THE TWO HOMEOSTATIC SYSTEMS
While the system that controls body water solute concentration (osmolality) – let us call it ADH/thirst system- rapidly responds (in a matter seconds to minutes) to small variations by setting in motion potent correcting mechanisms (changes in the drive for water ingestion and in the urine dilution or concentration) that finally return that concentrations to normal values within narrow fluctuation limits, the system that controls the volume of the extracellular fluid does so in a much slower way (in hours or days), is less precise and does not always return the volume to the value preceding the external challenge, settling for a limited stability that consists in only trying to match further gains with losses (I shall return to this point).
A small change in water balance (due to increased intake or exclusive water loss) translates into a change in osmolality (dilution or concentration), which, even if small, through the ADH/thirst system changes the behavioural and renal effector responses, eliminates or acquires water, and brings the body solute concentration back to the previous normal values. In this process it is not necessary to take into account the small variation in extracellular volume because its regulating system is practically not called into action.
Let us consider now, as an example, a sudden increase in the intake of hypertonic sodium chloride (salted chips ): there is a rapid rise in osmolality which leads, through the ADH/thirst system to a positive water balance and, soon, what persists is an isotonic rise in extracellular fluid volume, setting then in motion the volume control system. Even with excess hypotonic sodium chloride ingestion there will be a noticeable negative water balance, leaving as before an increase in isotonic extracellular fluid volume (albeit of a smaller magnitude) triggering the volume control system adaptation2.
As a mental framework, it is convenient to think in terms of two systems: consider water metabolism resulting in solute concentration and under the concentration regulation system supervision and, on the other side, salt metabolism resulting in extracellular fluid volume and under the volume regulation system supervision.
This is obviously an oversimplification and the two control systems are not completely independent.
It is important to consider the dual control (or, to be more precise, multifactorial control) of ADH secretion: under situations of marked hypovolemia there is an increase in ADH secretion and in thirst, resulting in some sacrifice of tonicity for the preservation of fluid volume. In situations of diminished effective arterial volume there is also a non-osmotic stimulus for ADH secretion- hyponatremia of congestive heart failure for instance. Again there are other known non-osmotic stimuli for ADH secretion, but keeping the list in mind (or readily available in electronic sources), the merits of the conceptual and didactic approach of always considering the two homeostatic systems cannot be overstated3.
VOLUME REGULATION
Due to the fact that sodium is by far the main extracellular cation and exists in a fixed concentration, its total body quantity determines the volume of the extracellular fluid. Under normal conditions the extracellular volume also has a strict correlation with the intravascular volume.
The system controlling volume is conceptually rather more complicated than the one controlling concentration. There are low-pressure sensors and high-pressure sensors, respectively in the venous and arterial sides of the circulation. The signals they transmit to the central integrator can be antagonistic (for instance augmented pressure in low-pressure baroreceptors and diminished pressure in high-pressure baroreceptors, in cardiac failure).
The general picture is even more complicated in reality, given the fact that the filling volume of vessels does not translate linearly and univocally into pressure: pressure is also determined by pump output, peripheral resistance and compliance of vessels, which can, by changing distensibility (mainly in veins), accommodate varying volumes without great pressure changes. Additionally, there are different behaviours in local vascular territories, with vasoconstriction in certain areas and vasodilation in others.
As a result, there is not, as yet, a unifying and comprehensive algorithm that can explain the regulation of sodium balance in normal physiology and in disease situations.
The pattern that seems to emerge is one of a slow and relatively imprecise control of sodium balance when the situation is dealing with small to moderate variations in intake or losses, mediated mainly by low-pressure baroreceptors as long as high-pressure sensing changes are not large. In these settings, a stable situation can be reached with the net balance becoming 0, even though the previous gain (or loss) persists if under 2 litres of extracellular fluid (with inter-individual variability).
How can one explain the behaviour of the control system in situations in which, retaining functionality, it allows the accumulation of large excess extracellular fluid (generalized oedema)? The common explanation is that what prevails among conflicting stimuli is the effective arterial blood volume, due to the fact that an adequate tissue perfusion better guarantees survival.
Effective arterial blood volume (EABV) is not measured directly by any sensor: even arterial baroreceptors, responding to vessel wall distension, closely related to intraluminal pressure, which in turn depends on blood flow, peripheral resistance and distensibility of the artery itself, do not measure EABV. EABV is more of a theoretical construct invoked to give coherence to the whole regulation system: when tissue perfusion is under menace (due to a low EABV), sodium retaining mechanisms are put to work even though the extracellular volume is already clearly expanded (oedema presence).
Situations like hepatic cirrhosis, nephrotic syndrome, congestive heart failure are the typical examples of the conflicts that may arise out of the messages sent by the various receptors of the volume control system and of how the central integration ends up being done, prevailing the interpretation of diminished EABV over an expanded extracellular volume.
There are known semiological difficulties in the evaluation of euvolaemia even with the aid of sophisticated auxiliary diagnostic means (isotopic dilution measures of total body water, vena cava size and distensibility). There are no normal limits to whatever is valuated as a measure of volaemia. Euvolaemia is a relative concept: it is the best value for that person in those circumstances. That explains the frequent need in clinical practice to the use of the tentative fluid challenge, a trial and error manoeuvre to exclude eventual undetected functional hypovolemia.
In all these situations the volume control system is fully operational and there is a trade-off optimization of circulatory and volume adjustments to respond to the pathological dysfunction (hypoalbuminaemia, portal hypertension, vasodilation in hepatic cirrhosis, for instance). More rarely it is the control system itself that is responsible for the pathological changes (autonomic dysfunction).
EXTRACELULAR VOLUME AND EFFECTIVE ARTERIAL BLOOD VOLUME
a) Compartments
The Na-K ATP dependent pumps located on the cell membranes transport Na out of cells and, inversely, K inside, making Na the main solute of the extracellular fluid and K the one of the intracellular compartment.
Water diffuses freely between those compartments. Water represents 50% (in women) to 60% (in men) of total body weight.
As we have seen, total body Na content determines the volume of extracellular fluid (ECF), 1/3 of total water under normal conditions.
The second barrier in body water is the capillary wall, dividing extracellular water in two compartments: interstitium and intravascular. The transport determinants at this interface are well described by Starling forces (hydrostatic and colloid-osmotic forces along the capillary wall). Interstitial volume is 3/4 of the extracellular volume while the intravascular is 1/4.
The volume of the extracellular fluid is kept within limits by homeostatic mechanisms which oppose disturbances resulting from variable intakes or losses.
In general, fluctuations in ECF volume run in parallel with intravascular volume except when there are changes in Starling forces (decreased oncotic pressure, for instance), or when, as referred above, keeping EABV overrides avoiding excess ECF volume (which seems like a regulation failure).
b) Regulation
Volume regulation can be approached considering an afferent limb with sensors collecting pressure data, central integration of that information by the nervous system, with a response consisting on an increase or decrease in sodium and water excretion mediated by an efferent limb intervening on kidneys and vasomotor tone.
Here is a short description of the two limbs and the resulting integration4.
Afferent limb
There are pressure/stretch sensors located in strategic places of the vasculature: low-pressure cardiovascular sensors (in the auricles, ventricles and pulmonary vessels), arterial high-pressure baroreceptors (carotid, aortic arch and renal vessels) and sensors in the central nervous system and in the liver vessels.
The existence of sensors in several territories allows for a differentiated monitoring: a sodium overload expands the ECF, situation that will be recognized through auricle distension, increasing the production of atrial natriuretic peptide (ANP) and leading to natriuresis by the latter action on the kidneys culminating in a new steady balance; a descent of systemic arterial pressure will be sensed by carotid baroreceptors leading to sympathetic activation (vasoconstriction and tendency for sodium retention), but the response may depend on whether the hypotension is caused by hypovolaemia (ex. haemorrhage) or cardiac failure, in which case the heightened ventricular pressure can dampen sodium retention through ANP or brain natriuretic peptide (BNP) secretion.
The feedback loops may be predominantly neural and local but often also involving e humoral components that may have systemic effects: juxtaglomerular apparatus sensing/modulating the secretion of renin; and changes in tubular flow and composition determining haemodynamic changes in glomerular circulation.
Efferent limb
Various mechanisms involving the renal handling of sodium (and also arteriolar tone) can be listed as effector responses in homeostatic constant volume preservation.
Sympathetic: sympathetic nerve endings reach afferent and efferent glomerular arteriole cells, as well as juxtaglomerular apparatus and renal tubule cells.
Sympathetic stimulation has a net sodium retention effect as well as selective vasoconstriction. These actions are direct and indirect. Renal nerves change the tone of afferent and efferent arterioles increasing filtration fraction (FF) which predisposes to sodium retention. They also directly stimulate proximal tubule cell sodium reabsorption and also the secretion of renin by the juxtaglomerular apparatus. This activation of the renin/angiotensin axis amplifies the direct effects on the kidney sodium handling.
Renin/Angiotensin/Aldosterone System: the liberation of renin, determined by sympathetic stimulation or through the juxtaglomerular apparatus leads to activation of angiotensin I, and by action of the angiotensin-converting enzyme to angiotensin II (Ang II). Ang II has a systemic vasoconstrictor effect but predominantly causes vasoconstriction of the efferent arteriole, acts directly on proximal tubule cells and, in positive feed-back loop increases sympathetic neuro-transmission. Additionally, it stimulates the production of aldosterone. All these actions converge in to increase sodium reabsorption.
Prostaglandins: PGI2 mediates the Renin production by the baroreceptor; PGE2, whose production is stimulated by Ang II, AVP and catecholamines, has a renal vasodilatory effect and inhibits water and salt reabsorption, dampening the renal effect of the very hormones that lead to its production, so, basically maintaining renal function (effect that is jeopardized when inhibited by the administration of non-steroidal anti-inflammatories).
AVP: As we shall see, the production of AVP, although strictly linked to the regulation of osmolality, is also stimulated by low circulatory volume. It has a vasoconstrictor effect (in high concentration – V1 receptors), and leads to water retention and increases sodium reabsorption and potassium excretion (partly in synergistic action with aldosterone) Natriuretic peptides: ANP (auricles) and BNP (ventricles) are peptides that stimulate diuresis, natriuresis and vascular muscle relaxation, through actions on the glomerulus, tubule and decreasing renin and aldosterone secretion; a similar peptide, CNP, produced by endothelial cells can have a role in local regulation.
Direct effect of blood pressure (Pressure Natriuresis): A rise in arterial pressure leads to a marked natriuretic response, even without the intervention of the usual neuro-humoral mediators (it seems to be mediated locally in the kidney by a rise in nitric oxide).
Integration
We do not have, as yet, an integrated vision of the whole operation of the several mechanisms to be able to build a satisfactory model3. How does occur the integration of the information which would lead by itself to opposing responses? How is the information from different territories integrated? Is there recruitment of effectors under greater disturbances? Is there a hierarchy of signal meanings and of responses?
It seems there is a complex integration with a logic of recruitment: when the first line adaptive response reveals itself to be insufficient then others are called into action in succession, eventually more primitive, coarser and less fine-tuned but eventually more powerful. Some proof for this comes from the observation of the escape phenomena (escape from diuretic action, escape from aldosterone) and also the pressure-natriuresis data- when one experimentally fixes renin-aldosterone system in either low or high levels, it is then through the rise in blood pressure (renal perfusion pressure) that natriuresis follows the rise in salt intake and balance is reached. Facing persistent alterations away from equilibrium the subtler mechanisms are overridden by a stronger, simpler one (perfusion pressure itself) that creates a new 0 balance situation, although with different operational parameters – it takes a higher blood pressure to excrete the exceeding salt.
The complete hierarchy of of recruitment is not known. We only recognize general patterns.
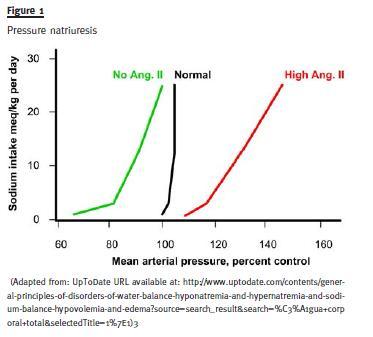
Apparently small, day-to-day changes in sodium intake or loss are dealt with by the actions on collecting tubules of varying levels of ANP or aldosterone, inducing the renal handling of sodium in the opposite direction of the detected change, correcting the disturbance and bringing body composition back to basal values.
When the disturbance is more severe or sudden, additional mechanisms are recruited with increasing levels of sympathetic activation and when the problem arises in the effector neuro-hormonal limb itself, there can be a homeostatic response, determined solely and directly by the arterial perfusion pressure on the kidneys (autoregulation).
c) Clinical setting
The clinical situations relating to extracellular volume are naturally dominated by volume contraction or expansion. It must be stressed out again that the clinical evaluation of the patient´s volemia is neither easy nor rigorous in situations of small deviations from normal/ideal values.
In volume depletion it is usual to start by trying to ascertain if the cause is renal (renal structures like tubules or effector deficiency) or if, by contrast, the losses are extra-renal (and the kidney is either actuallyreacting in an homeostatically adequate way or, by contrast its capacity has been overwhelmed).
In situations of volume overload it is also customary to start by trying to find out if sodium retention is primarily renal (renal disease) or if it is a secondary response to a decrease in effective arterial blood volume (cardiac failure, hepatic failure or nephrotic syndrome, mainly).
OSMOLALITY-NATREMIA
a) Compartments; typical balance
Osmolality depends on the number of free molecules in a certain amount of solvent. It is usually calculated as twice sodium concentration (it is assumed anions equal cations, so twice the concentration of the major cation is used) plus the concentration of glucose molecules (glucose in mg divided by its molecular weight-18) plus the molecular concentration of urea (concentration of urea divided by 6). This value would be identical to the reading obtained directly in an osmometer. Bigger molecules represent a small number of particles due to their large molecular weight and have a usually ignored impact.
We have seen that water diffuses freely through cell membranes, moving to the side with the higher tonicity and until, by the ensuing dilution, concentrations become equal and net water movement stops.
Urea and glucose have different biological behaviours, however: urea passes almost freely the membranes of most cells, its concentration is the same intra-and extracellularly and so it exerts no osmotic effect. As to glucose, it does not cross cell membranes so easily and so it does exert an osmotic effect, driving water out of cells when it rises markedly in extracellular fluid, inducing some decrease in cell volume and dilution of the components of the extracellular fluid, namely sodium, with resulting hyponatremia.
In urine the main osmols are the electrolytes (sodium, potassium and chloride), ammonia and urea.
Urea is one of the main determinants of urine osmolality and, contrary to what happens in the rest of the organism, in the kidney it exerts a marked osmotic effect due to varying urea and water permeabilities along the nephron. The amount of daily osmols eliminated by the kidney to keep 0 net balance depends on the diet, basically the amount of salts and protein ingested.
Changes in extracellular fluid tonicity determine variations in cell size by the influx or outflux of water down the osmotic gradient. Central nervous system cells being contained inside a rigid skull may suffer from acute changes in size and there are protective mechanisms to generate or get rid of intracellular osmols, although slow to develop.
Under typical conditions water drinking will range from 400 ml (obligatory minimum) to 1400 ml; 850 ml from water contained in foods will be added to intake; 350 ml of water from the oxidation of food in metabolic processes is also added; the body then receives a total of 1600 to 2600 ml of water a day.
In the same time period the water output is 500ml (minimum) to 1500 ml in urine; 500 ml in sweating/perspiration; 400 ml through respiration in expired saturated air; and about 200 ml in faeces. The organism is in balance. It is obvious that these are usual values. Skin water loss can increase markedly with heat or in fever; water in faeces can increase in diarrhoea. The minimum volume of daily dieresis (under which oliguria is present) is determined by the maximal concentrating capacity reachable by the kidneys (1400 mOsm/Kg) to eliminate 700 to 800 mOsm generated by a normal diet. Usually most people eliminate these osmols in a much more diluted urine.
b) Regulation
Tonicity of the extracellular fluid is actively kept within very narrow limits, around 288 mOsm/L, through the variation in daily water output in urine (from 500 ml to 12 000 ml) and also through induction of appropriate water drinking behaviour (thirst or lack of it). It is remarkable thirst can be such a strong motivation, becoming, in extreme situations, the sole determinant of behaviour and completely filling the field of consciousness!
Osmoreceptors in the hypothalamus control thirst and the secretion of ADH, the effector limb, which, acting on the kidney is responsible for varying urinary dilution.
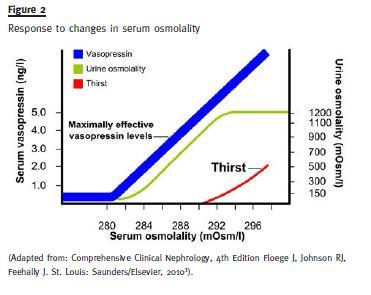
As can be seen in Figure 2, regulation is very tight: below 280 mOsm/L of plasma osmolality ADH is not secreted, there is no thirst and urine is dilute (minimum osmolality 40 to 100 mOsm/L); a little above 280 ADH starts being secreted in rising amounts and urine osmolality also rises gradually; when urine is isotonic (300 mOsm/L) one can be sure ADH is being secreted; at 290 mOsm/L thirst sets in and urine becomes increasingly concentrated (maximum value 1200 mOsm/L.
Due to the fact that, mainly in the ascending limb of Henles loop, sodium is reabsorbed from the tubular fluid not accompanied by water, hypotonic (intratubular) and hypertonic (interstitium) fluids are generated (the effect is amplified by counter current multiplication mechanisms). The intratubular hypotonic fluid can be eliminated as such if the remaining tubule is kept impermeable to water, or it can be concentrated by water outflowing towards the hypertonic interstitium if the tubule is rendered permeable to water through the action of ADH.
It is clear that reaching extremes of maximal concentration or dilution depends on additional factors, namely intratubular flow (which varies with ECF volume), the solute load to excrete (protein and solute poor diets hamper attaining maximal values). And it obviously depends on the structural and functional integrity of the control system and kidney.
The ADH secretion is not solely dependent on tonicity, it also depends on efective arterial blood volume and rises when the latter is diminished, leading to water conservation which tends to dampen volume contraction but at the price of a lowered tonicity (hyponatremia). As we have seen contraction of EABV can occur with a truly diminished ECF volume (salt losses) or with an expanded ECF (hyponatremia of heart failure). One must consider other independent stimuli to ADH secretion: pain, anxiety, several drugs and Ang II itself.
To quantify the degree of urine dilution or concentration we can use some useful concepts, like free-water clearance (or reabsorption) and electrolytefree water reabsorption. Basically, produced urine is considered as being divisible in two parts: one volume, containing all the solutes (osmols) in isotonic concentration with the plasma, and another volume of solute free water. If the urine is diluted then the volume of free water is positive and can be thought off as the amount of water that is being removed from the ECF. On the contrary, if the urine is concentrated regarding the plasma, the volume of free water is negative (reabsorption of free water) and the volume of free water equals the amount of water that is being added to the ECF.
As we have seen, urine osmolality depends not only on electrolytes but also on urea and ammonia.
As the prime determinants of plasma osmolality are its ions, one may wish to focus on the effects of urine water on the concentration of the ions. We then measure electrolyte-free water reabsorption and consider real urine divided into two parts: one that has all the electrolytes in a concentration equal to plasma and another volume of electrolyte-free water.
Again if the value is positive, it means this volume is being added to the plasma, this time diluting sodium (urinary urea and ammonia that are present independently of the electrolyte content of the urine are left out of the calculation).
c) Clinical setting
We propose a structured approach to the analysis of disturbances of ionic equilibrium, going through several steps in succession: Try to establish if the disturbance to homeostasis is recent and still challenging the adaptive mechanisms (induction phase) or if, on the contrary, a steady state (or balance) has been reached even at values somewhat far from normal.
Evaluate carefully ECF volume and try to define the situation as volume depletion, volume overload or grossly normal. If there is ECF volume expansion, try to characterize the situation in terms of the EABV, bearing in mind the situations where there is a decrease in this latter parameter (cardiac failure, nephrotic syndrome and liver cirrhosis) Only then consider natremia, as its meaning is completely different in the different clinical settings already outlined in the two previous steps (the same value of hyponatremia would have completely different clinical meaning in the setting of volume contraction yet in induction phase or in normal volume, in balance).
This approach is the basis for a rational, physiology-based search for the correct choice of tests and final diagnosis.
References
1. Floege J, Johnson RJ, Feehally J. Comprehensive Clinical Nephrology, 4th Edition. St. Louis, MO.: Saunders-Elsevier, 2010. [ Links ]
2. Halperin, ML, Kamel, KS, Goldstein MB. Fluid, Electrolyte and Acid-Base Physiology – A Problem-Based Approach, 4th Edition. Philadelphia, PA. Saunders-Elsevier, 2010. [ Links ]
3. Rose, Burton David, Post, Theodore W. Clinical Physiology of Acid-Base and Electrolyte Disorders, 5th Edition. New York, N.Y.: McGraw-Hill, 2001. [ Links ]
4. Schrier, Robert W. Renal and Electrolyte Disorders, 7th Edition. Philadelphia, PA.: Lippincott Williams & Wilkins, [ Links ] 2010
Dr. Pedro Correia
Department of Nephrology
Hospital Fernando da Fonseca, EPE
IC19, 2720-276 Amadora, Portugal
E-mail: pedrocorreia@me.com
Conflict of interest statement: None declared.
Received for publication: 9/02/2015
Accepted in revised form: 23/02/2015

