










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.29 no.1 Lisboa mar. 2015
CASE REPORT
Idiopathic hypocomplementaemic tubulointerstitial nephritis
Nefrite túbulo-intersticial hipocomplementémica idiopática
Ana Azevedo1, Isabel Mesquita1, Helena Viana1, Gizela Rocha2, Carina Ferreira1, Vasco Fernandes1, Antonio Murinello2, Fernanda Carvalho1, Fernando Nolasco2
1Department of Nephrology, Hospital Curry Cabral, Centro Hospitalar de Lisboa Central. Lisboa, Portugal 2Department of Medicine 1, Hospital Curry Cabral, Centro Hospitalar de Lisboa Central. Lisboa, Portugal
ABSTRACT
Background: Tubulointerstitial nephritis (TIN) is a common cause of kidney injury typically seen in association with drug exposure, infection or autoimmune diseases. However, TIN with interstitial immune complex deposition, without glomerular injury, is rarely observed. Case: We report a case of a 64-yearold Indian woman admitted for dialysis-requiring renal failure, without involvement of other organs. Urinalysis showed blood 3+ and 24h proteinuria of 1.5 g. Renal ultrasound revealed normal sized kidneys with loss of parenchymal-sinus differentiation. Laboratory tests disclosed low C3, positive ANA but negative anti-dsDNA, SSA and SSB. Serum protein electrophoresis was normal. The renal biopsy showed tubulointerstitial nephritis with positive immunoglobulin staining involving the interstitium and tubular basement membrane with glomerular sparing. The patient started prednisolone (1mg/kg/day) without recovery of the renal function. Conclusion: Idiopathic hypocomplementaemic tubulointerstitial nephritis is a rare disease with few cases described in the literature. To our knowledge this is the first case reported in Portugal.Key-Words: Hypocomplementaemia; idiopathic tubulointersticial nephritis; renal failure.
RESUMO
A nefrite tubulo-intersticial (NTI) é uma causa comum de lesão renal, tipicamente associada a toma de fármacos, infecções ou doenças auto-imunes. No entanto, as NTI com deposição de imuno-complexos no interstício, sem envolvimento glomerular são raramente observadas. Apresentámos o caso de uma doente do sexo feminino, 64 anos, etnia indiana, admitida no nosso hospital, por insuficiência renal dependente de diálise. A análise sumária de urina revelou hematúria (3+) e a proteinúria nas 24h foi quantificada em 1,5g. A ecografia renal revelou rins de dimensões normais com perda da diferenciação parenquimo-sinusal. Analiticamente salienta-se, C3 baixo, ANA positivo, mas anticorpo anti dsDNA, SSA e SSB negativo. A electroforese de proteínas séricas não apresentava alterações. O doseamento de imunoglobulinas era normal, sem evidência de componente monoclonal. O restante estudo imunológico não apresentou alterações. Durante o internamento a doente realizou uma biópsia renal que mostrou nefrite tubulo-intersticial, com depósito de imunoglobulinas envolvendo o interstício e a membrana basal tubular, poupando os glomérulos. A doente iniciou terapêutica com prednisolona (1mg/Kg/dia), sem recuperação da função renal. A nefrite tubulo-intersticial hipocomplementémica idiopática é uma doença rara, com poucos casos descritos na literatura. Tanto quanto sabemos, este é o primeiro caso descrito em Portugal.
Palavras-Chave: Hipocomplementémia; insuficiência renal; nefrite túbulo-intersticial idiopática.
INTRODUCTION
Tubulointerstitial nephritis is classified into allergic, autoimmune, infectious and idiopathic1. Most forms of interstitial nephritis are cell mediated, with inflammatory infiltrates in interstitium without tubulointerstitial immune deposits2. Interstitial nephritis with tubulointerstitial immune deposits and glomerular sparing can occur in Sjögren disease, lupus nephritisand in other two recently described pathologies: IgG4-related systemic disease and idiopathic hypocomplementaemic tubulointerstitial nephritis3.
Thirteen cases of idiopathic hypocomplementaemic tubulointerstitial nephritis have been described1,2,4-7.
They were primarily characterized by tubulointerstitial immunological impairment and most patients were older men.
CASE REPORT
A 64-year-old Indo-Caucasian woman, presented to our emergency room with lower limb oedema and dyspnoea for minimal exertion, with 2 weeks of evolution and progressive worsening in the previous week. She also reported nocturia in the previous 3 months and a progressive decrease in urine output in the last few days.
There was no history of arthralgia, alopecia, oral ulcers, Sicca syndrome, skin disorders, allergies, haematuria, foam urine, dysuria, fever, weight loss, nephrolithiasis, recurrent urinary tract infection, taking of NSAIDs, dietary supplements, natural medicines, antibiotics. She also denied animal contact and recent trips. The patient was unaware of previous impaired renal function and she denied other pathologies.
The patient had been submitted to a radical hysterectomy, in 2008, due to a cervical carcinoma in situ and uterine myoma. She was kept under regular surveillance, without evidence of worsening. The patient related smoking and light alcohol consumption habits in the past.
On admission, she was conscious, oriented, pale, afebrile and hydrated. She presented polypnoea, jugular engorgement (+++), inspiratory rales at both lung bases and peripheral oedema (+). Her blood pressure was 165/86mmHg. The remainder of her physical examination was unremarkable.
Initial laboratory testing results revealed abnormal levels of serum creatinine – 11.85mg/dl (GFR 4ml/min/1.73m2, calculated by CKD-EPI creatinine equation), urea – 205mg/dl, potassium – 5.4mmol/l, calcium – 6.2mg/dl, phosphorous – 10.3mg/dl, albumin – 2.5g/l.
Serum levels of sodium, creatinine phosphokinase (CPK) and lactate dehydrogenase (LDH) were normal. The haemogram presented: haemoglobin – 8.0g/dl (normocytic, normochromic), platelets – 254x10^3mm3, leukocytes – 5x10^9/L; (neutrophil 73.4%, lymphocytes 21.5%, monocytes 1.8%, eosinophils 2.5%, basophil 0.8%).
Arterial blood gases revealed a metabolic acidaemia: pH – 7.31 and HCO3 – 12mmol/l. Urinalysis showed haematuria and proteinuria (protein- 300mg/dL; blood-3+cells/HPF), many epithelial cells and no casts.
Renal ultrasound revealed normal sized kidneys with parenchymal-sinus differentiation loss.
The patient started haemodialysis through a tunnelized catheter and she was hospitalized for diagnostic and treatment guidance.
Subsequent laboratory study disclosed: low C3 [55.2mg/dL (N = 75.0-140.0mg/dl)], normal C4, positive ANA > 1:160 with fine granular pattern, angiotensin-converting-enzyme (ACE) 117U/L (N = 8-52 U/L) and iPTH of 841 pg/ml (N = 10-70). The remaining laboratory tests, including anti-dsDNA, anti-GBM, ANCA, rheumatoid factor, anti-Jo 1, anti-SSB/La, anti-SSA/Ro, HBsAg, anti-HCV, anti-HIV and protein electrophoresis, were negative. The serum immunoglobulins IgA, IgM and IgG were normal and there was no evidence of monoclonal component. The urinary immunofixation was unremarkable. Thyroid function tests were normal. The 24h proteinuria was 1.5g.
Complementary studies: chest X-rays, ECG and 2D-echocardiogram did not reveal significant changes with the exception of a small pleural and pericardial effusion. The chest computed tomography excluded hilar lymphadenopathy. Bronchoscopy with bronchoalveolar lavage did not show CD4/CD8 inversion.
The performed ophthalmologic observation excluded granulomas, infection, uveitis and choroiditis.
Renal biopsy was performed and a total of 19 glomeruli were obtained. Five glomeruli showed global sclerosis and six glomeruli showed segmental sclerosis lesions. The remaining glomeruli showed hypertrophy/hyperplasia of podocytes and Bowmans capsule thickening (Fig. 1 and Fig. 2).
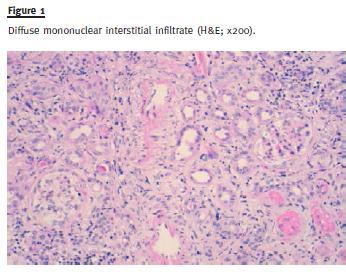
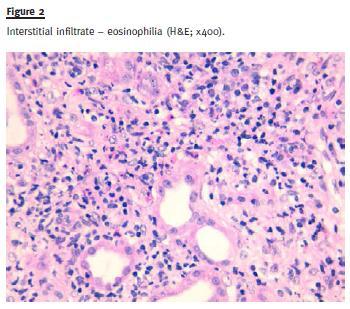
Large areas (> 90% of the sample) of interstitial fibrosis and tubular atrophy were observed with marked inflammatory infiltrate composed of mature lymphocytes, plasma cells and occasional neutrophils and eosinophils. No granuloma was observed (Fig. 3).
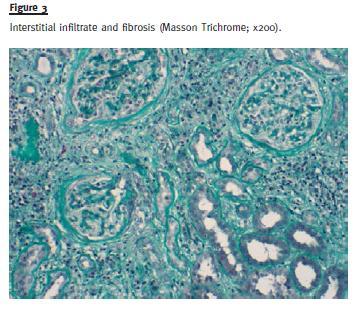
There was fibrous thickening of arterial intima in medium size arteries without arteriolar hyalinosis.
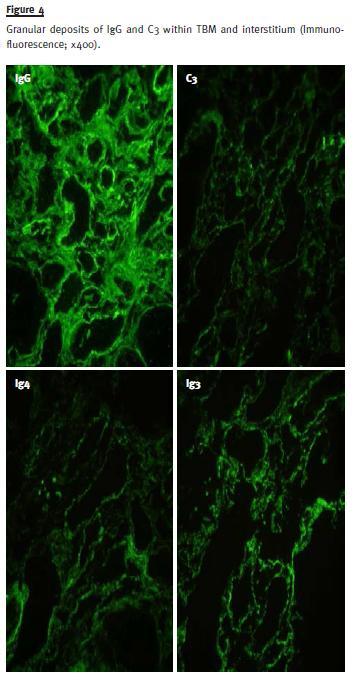
Immunofluorescence in frozen section demonstrated granular deposits in the interstitium and tubular basal membrane of IgG (++), C3 (++), C1q (++/+++), κ chain (+), IgG1 (++), IgG2 (++), IgG3 (+++) and IgG4 (++) (Fig. 4).
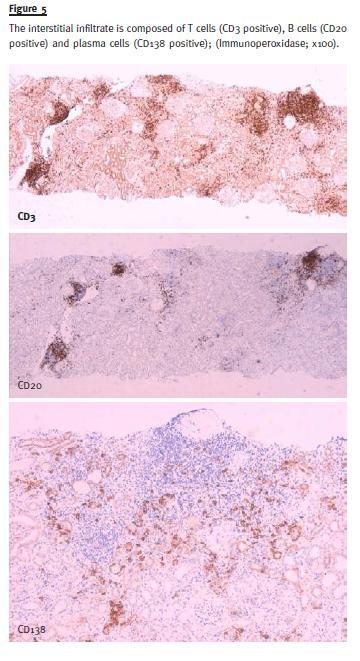
Characterization of infiltrate was made by immunohistochemistry in paraffin section and revealed CD3 (+++), CD138 (++) and CD20 (+) (Fig. 5).
We diagnosed this case as hypocomplementaemic idiopathic tubulointerstitial nephritis, and prednisolone 1mg/kg/day (40 mg/day) was started. The patient was discharged 3 weeks after without renal function recovery. She remained on chronic haemodialysis programme and she had been followedup since discharge in the ambulatory clinic. She continued therapy with prednisone 40mg/day for 4 months, without recovery of the renal function.
However there was normalization of C3 levels (124mg/dL).
The patient was re-hospitalized, after this period, due to fever of unknown origin, associated with myalgia and tiredness. After aetiologic research, miliary tuberculosis was diagnosed. The patient began therapy with a 4-drug regimen (isoniazid, rifampin, pyrazinamide and ethambutol).
During her stay in hospital, she suffered several healthcare associated infections, after which she suffered multi-organ failure, reason why the patient was transferred to an Intensive Care Unit. Here she needed invasive mechanical ventilation and vasoactive amines, and death occurred several days later.
DISCUSSION
We present a case of a patient with renal failure, hypocomplementaemia (low C3), positive ANA, active urine sediment (blood 3+ cells/HPF) and 24h proteinuria of 1.5g. She did not have eosinophilia and anti-dsDNA, SSA and SSB were negative. The biopsy revealed tubulointerstitial immunological damage with C3 and all IgG subtypes deposition in the tubular basement membrane (TBM) and interstitium, in a granular and diffuse pattern without glomerular involvement. We diagnosed this case as hypocomplementaemic idiopathic tubulointerstitial nephritis, a very rare cause of tubulointerstitial nephritis1.
Tubulointerstitial nephritis is related to various causative factors, including infectious, allergic, toxic, autoimmune and idiopathic conditions2,8. Linear or granular TBM deposits may be found in systemic lupus erythematosus (SLE), Goodpastures syndrome, Sjogrens syndrome (SS), and membranous glomerulopathy, and rarely, usually with linear pattern, in drug-induced nephropathy secondary to methicillin and phenytoin1,2,4,5,9. It has also been described in urticarial vasculitis syndrome, adenovirus infection, renal allograft rejection or mixed cryoglobulinaemia4.
The majority of these diseases have predominantly glomerular involvement, which was not present in our case. Predominant or isolated granular tubulointerstitial deposits are usually restricted to autoimmune disorders, such as SLE, SS and IgG4-related systemic disease.
The SLE was excluded as the patient did not fulfill the American Rheumatology Association diagnostic criteria10.
Negative serology for anti-SSA/Ro and anti-SSB/La and no symptoms or signs suggestive of SS ruled out this entity11.
The IgG4-related disease is a clinicopathological entity characterized by tumefactive lesions, a dense lymphoplasmocytic infiltrate rich in IgG4-positive plasma cells with fibrosis, and elevated total IgG or IgG4 in serum. In the kidney, the most dominant feature is a tubulointerstitial nephritis3. A Japanese group proposed a diagnostic algorithm for IgG4-related kidney disease (IgG4 RKD)12. In accordance with this algorithm, IgG4 RKD should be suspected when some kidney damage is present associated to an elevated serum total IgG level, hypocomplementaemia or an elevated IgE level. Other characteristics that help in the diagnosis of this entity are serum IgG4 level (> 135mg/dL), radiologic findings like diffuse kidney enlargement, and a renal biopsy with evidence of plasma cell-rich TIN with > 10 IgG4+plasma cells/HPF fill in the most concentrated field.
Using this diagnostic algorithm for IgG4 RKD, we ruled out this hypothesis.
Only thirteen cases1,2,4-7 of idiopathic hypocomplementaemic interstitial nephritis have been described in the literature. The largest series is that by Kambham et al. with eight cases reported2.
The significant varied nature between the reported cases challenged the possibility of a common aetiology1,4, but kidney biopsies of the reported cases always showed TBM deposits with granular and predominantly diffuse staining for IgG. Hypocomplementaemia, TBM immune complexes and glomerular sparing were consistent findings in all cases1,2,4,5.
This indicates that the immune deposits in the TBM are capable of activating serum complement, leading to immune-mediated tubular injury1. C3 staining in the TBM was not, however, a consistent finding among the 13 cases, as also IgM, C1q, Lambda and kappa chains1,2,4-7. The pathogenesis of TBM deposits is unknown. The TBM antibody–antigen complex could result from either in situ immune complex formation or deposition of preformed circulating immune complexes1,5.
Tubulointerstitial nephritis with immune deposits seems to occur predominantly in older men with underlying immunologic disorders, including sclerosing cholangitis (two cases), marginal B cell lymphoma (one case), leucocytoclastic vasculitis (one case)2.
Adult onset of diabetes mellitus and coronary artery disease were also present in five and six, respectively1,2,6,7.
However, in our case the patient was a woman and she did not present any known immunologic condition.
Although experience in treating idiopathic hypocomplementaemic tubulointerstitial nephritis is strictly limited because of the few cases, the described ones were all treated with immunosuppressant drugs2,6,7 including, at least, prednisone. Renal function (reflected by serum creatinine) improved in 9/13 cases (significant in 6 patients)1,2,4,5. Prednisone doses were not described in all reported cases but it seems that 1 mg/kg/day was consistent in more recent cases1,4,5.
It is unlikely that the remission noted in these patients was related to a particular immunosuppressant, because similar regimens were used in patients in whom there was no clinical improvement.
The variability of treatment response indicates the potentially progressive nature of this disease1,5 and could be related with the presence of irreversible lesions/different degrees of chronicity4, although Vaseemuddin et al support that there is no apparent correlation between biopsy findings and clinical progression1.
Even though, so far the few reports support an early5 and prolonged therapy1,4 with a median of 8.5 months (3-20 months)4 that at least includes prednisone.
In the scenario of treatment failure, additional immunosuppressants could be associated1,4. In four cases, further immunosuppressant agents were added: in two cyclophosphamide, one tacrolimus plus mycophenolate mofetil and in the last case mycophenolic acid. The patients that have done cyclophosphamide, one had a marginal B cell lymphoma, and another developed thrombotic thrombocytopenic purpura requiring plasmapheresis1. The third patient had cirrhosis and a monoclonal interstitial infiltrate and underwent liver transplantation several weeks after the biopsy, and was thereafter maintained on tacrolimus, mycophenolate mofetil and prednisone1.
The last patient started mycophenolic acid after hyperglycaemic development allowing prednisone withdrawl and subsequent glycaemic control4. Mycophenolate mofetil has been reported as a useful therapeutic option for steroid-resistant acute interstitial nephritis (AIN) and may be considered as potential first-line therapy in selected patients4.
There are no known indicators of disease progression or treatment response, but two prior cases1,4 have revealed normalization of the complement level as renal function enhanced. Hypocomplementaemia was suggested as a marker of clinical activity4.
In our case, given the patient history of cancer and the presence of large interstitial fibrosis and tubular atrophy, we choose to treat her only with corticosteroids, not associating any other immunosuppressor.
We decided to extend therapy, because some similar cases with severe interstitial fibrosis and tubular atrophy have been published in which, after prolonged treatment with corticosteroids (3-9months), there was recovery of the renal function1,2.
Despite previous publications defined IgG4-related disease as being a systemic condition described in virtually every organ system1,4,5,9,10, Stone et al. (2012) considered that many medical conditions that have long been viewed as disorders confined to single organs are part of the spectrum of IgG4-related disease. One of these suggested conditions is idiopathic hypocomplementaemic tubulointerstitial nephritis with extensive tubulointerstitial deposits13.
Before Stone, Raissian et al. had already suggested that many patients diagnosed with idiopathic hypocomplementaemic tubulointerstitial nephritis likely represent IgG4-related disease, especially those cases with associated immunologic conditions3. However, some diagnostic criteria to the IgG4 related disease12 have recently been published, which allow a higher homogeneity classifying this entity.
In summary, idiopathic hypocomplementaemic tubulointerstitial nephritis is differential diagnosis of renal failure and hypocomplementaemia, has a progressive nature and requires an early diagnosis and prolonged therapy.
References
1. Vaseemuddin M, Schwartz MM, Dunea G, Kraus MA. Idiopathic hypocomplementemic immune-complex-mediated tubulointerstitial nephritis. Nat Clin Pract Nephrol 2007; 3(1): 50–58. [ Links ]
2. Kambham N, Markowitz GS, Tanji N, Mansukhani MM, Orazi A, DAgati VD. Idiopathic hypocomplementemic interstitial nephritis with extensive tubulointerstitial deposits. Am J Kidney Dis 2001; 37:(2):388–399. [ Links ]
3. Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011; 22(7):1343-1352. [ Links ]
4. Olea T, Picazo ML, Martínez Ara J, Díaz Mancebo R, Riñón C, Selgas R. Idiopathic hypocomplementemic interstitial nephritis treated with mycophenolic acid. Port J Nephrol Hypert 2011; 25(1): 47-50. [ Links ]
5. Gupta A, Jothy S, Somerville P, Zaltzman JS. Hypocomplementaemic immune complex tubulointerstitial nephritis. NDT Plus 2010;3(1): 78–80. [ Links ]
6. Hyun J, Galen MA. Acute interstitial nephritis. A case characterized by increase in serum IgG, IgM, and IgE concentrations. Eosinophilia, and IgE deposition in renal tubules. Arch Intern Med 1981; 141(5):679–681. [ Links ]
7. Tokumoto M, Fukuda K, Shinozaki M, et al. Acute interstitial nephritis with immune complex deposition and MHC class II antigen presentation along the tubular basement membrane. Nephrol Dial Transplant 1999; 14(9):2210–2215. [ Links ]
8. Cavallo T: Tubulointerstitial nephritis, in Jennette JC, Olson JL, Schwartz MM, Silva FG (eds): Heptinstalls Pathology of the Kidney (ed 5). Philadelphia, Lippincott-Raven, 1998, pp 667-723. [ Links ]
9. Lehman DH, Wilson CB, Dixon FJ. Extraglomerular immunoglobulin deposits in human nephritis. Am J Med 1975;58(6):765 -796. [ Links ]
10. Mori Y, Kishimoto N, Yamahara H, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus nephritis. Clin Exp Nephrol 2005; 9(1): 79–84. [ Links ]
11. Goules A, Masouridi S, Tzioufas AG, Ioannidis JP, Skopouli FN, Moutsopoulos HM. Clinically significant and biopsy-documented renal involvement in primary Sjögren syndrome. Medicine (Baltimore) 2000; 79(4): 241–249. [ Links ]
12. Saeki T, Kwano M. IgG4-related kidney disease. Kidney Int 2014; 85(2):251-257. [ Links ]
13. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366(6):539–551. [ Links ]
Ana Azevedo
Department of Nephrology, Hospital Curry Cabral, Centro Hospitalar de Lisboa Central,
Rua da Beneficiência, nº 8; 1069-166 Lisboa, Portugal.
E-mail: azevedo.gomes.ana@gmail.com
Conflict of interest statement: None declared.
Received for publication: 08/09/2014
Accepted in revised form: 13/01/2015

