










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.29 no.3 Lisboa set. 2015
CASE REPORT
Antiphospholipid syndrome and nephrotic syndrome: are they related?
Síndrome do anticorpo antifosfolipídico e síndrome nefrótica: estarão relacionados?
1Alice Lança, 1Rachele Escoli, 1Francisco Ferrer, 1Sequeira Andrade
1Nephrology Department, Centro Hospitalar Médio Tejo, Torres Novas, Portugal.
ABSTRACT
The antiphospholipid syndrome is a clinical entity that presents as arterial or venous thrombotic events in association with circulating phospholipid autoantibodies. Patients may display a constellation of neurologic, cardiovascular, obstetric and renal complications as a result of antibody-induced vessel injury. Kidneys are one of the most affected organs and its involvement often reflects the extension of the thrombotic process in its vasculature. However, in addition to the characteristic vascular findings of antiphospholipid nephropathy, an expanding spectrum of renal lesions has been reported. Primary glo- merulopathies may often overcome the clinical picture and present themselves as nephrotic syndrome with or without renal dysfunction. Here, we report a case of a 45-year old woman with antiphospholipid syndrome that presented to the emergency department with generalized oedema. After the initial workup nephrotic syndrome was assumed and a renal biopsy was performed. No detectable glomerular abnormalities other than sclerosis as a result of subcapsular ischaemia were seen. Immunofluorescence was negative for glomeruli deposits. The paucity of lesions in the non-sclerosed glomeruli pointed a primary podocytopathy as the most probable cause. The patient was started on corticosteroids with resultant complete remission in about 8 weeks. Nonetheless, during steroid tapering, the nephrotic syndrome relapsed and a higher dose was again needed for remission. However, the patient had to remain on low-dose corticosteroids for maintenance of nephrotic syndrome regression. Its sudden onset, together with the histological features and the good steroid response, made minimal change disease the most probable diagnosis. Yet, it remains unclear whether minimal change disease was related with antiphospholipid syndrome or was an unfortunate event.
Key-Words: Antiphospholipid syndrome; minimal change disease; nephrotic syndrome.
RESUMO
A síndrome do anticorpo antifosfolipídico é uma patologia que se manifesta pela presença de eventos trombóticos arteriais ou venosos associados a autoanticorpos antifosfolípidicos em circulação. As lesões vasculares induzidas pelos anticorpos resultam numa constelação de complicações neurológicas, cardiovasculares, obstétricas e renais. O envolvimento renal é frequente e reflecte a extensão do processo trombótico nas vascularização renal. Contudo, para além das lesões vasculares características da nefropatia desta síndrome, várias outras alterações renais têm sido relatadas. As glomerulopatias primárias podem sobrepor-se ao quadro clínico característico desta nefropatia e apresentar-se como síndrome nefrótica com ou sem disfunção renal. O caso descrito corresponde a uma mulher de 45 anos com síndrome do anticorpo antifosfolipídico, que recorreu ao Serviço de Urgência por quadro de edemas generalizados. Após a avaliação inicial, perante o diagnóstico de síndrome nefrótica realizou-se biópsia renal. Não se visualizaram alterações glomerulares além da esclerose associada a isquémia subcapsular. A imunofluorescência foi negativa para depósitos glomerulares. A escassez de lesões nos glomérulos não esclerosados sugeriu a podocitopatia primária como a causa mais provável da síndrome nefrótica, pelo que iniciou corticoterapia com resultante remissão completa ao fim de 8 semanas. No entanto, durante a redução progressiva da terapêutica verificou-se recaída desta síndrome com necessidade de aumentar a dose de prednisolona. Porém, constatou-se que a manutenção da regressão da síndrome nefrótica só foi possível com a toma permanente de corticoterapia em baixas doses. O início súbito do quadro, juntamente com as características histológicas e a boa resposta à corticoterapia, apontou a doença de lesões mínimas como o diagnóstico mais provável. Todavia, permanece ainda por esclarecer se esta glomerulopatia está relacionada com a síndrome anticorpo antifosfolípido ou se resultou apenas de um acaso infeliz.
Palavras-Chave: Doença de lesões mínimas; síndrome do anticorpo antifosfolipídico; síndrome nefrótica.
INTRODUCTION
The antiphospholipid syndrome (APS) is an autoimmune disorder that leads to a prothrombic state in the presence of circulating phospholipid autoantibodies (APL) in otherwise healthy patients. It can occur isolated (primary APS) or in association with other systemic diseases (secondary APS) such as systemic lupus erythematosus (SLE) and less com- monly rheumatoid arthritis, systemic sclerosis, and primary Sjögrens syndrome1,2. It has also been described as occurring associated with a primary glomerulopathy, such as membranous nephropathy, minimal change disease (MCD) or pauci-immune anti- neutrophilic cytoplasmic antibody (ANCA)-negative nephritis3. A more serious, often fatal, form of disease is called catastrophic antiphospholipid antibody syn- drome (CAPS) characterized by multiple organ infarctions1,4. According to the updated 2006 criteria for APS, it is diagnosed by one clinical criterion (thrombosis, pregnancy morbidity) and one laboratory criterion (anticardiolipin, anti-ß2-glycoprotein or lupic anticoagulant) two or more occasions at least 12 weeks apart)3-5. The clinical manifestations are generally seen in patients between 35 and 45 years of age and, despite mainly associated with thrombotic lesions, they also can result from pathological features distinct from vascular injury. Usually patients develop obstetrical complications (spontaneous abor- tions, stillbirth and severe eclampsia), recurrent ischaemic strokes, deep venous thrombosis, pulmonary embolism and livedo reticularis5. Renal involvement represents one of the most important features of this syndrome. It results from renal vasculature occlusion due to thrombi formation (APS nephropathy, APSN) and its clinical expression may vary from mild to heavy proteinuria [although nephrotic syndrome (NS) is rare], microscopic haematuria, hypertension and rapidly progressive renal failure2. Histopathological findings usually show ischaemic glomeruli and thrombotic lesions, without glomerular or arterial immune deposits on immunofluorescence. Subcapsular cortical atrophy and interstitial fibrosis among areas of normal parenchyma is considered to be very typical of APSN6. However, when there is a primary glomerulopathy associated, the glomerular abnormalities may prevail over the vascular lesions and the clinical picture may arise as NS2.
CASE REPORT
A 45-year-old Caucasian female was admitted to the hospital with progressive generalized oedema and decreased urinary output over the previous month. The patient had a personal history of two spontaneous miscarriages and arterial hypertension which was detected after an ischaemic stroke 10 years earlier. At that time, she was diagnosed with primary APS and was started on warfarin 5mg 1id, Lisinopril 20 mg 1id, atorvastatin 10 mg 1id. Also, she underwent major depression for which she was medicated with quetiapine 100 mg 1id and moclobemide 150 mg 1id. The follow-up since then was uneventful until 1 month prior to admission when she developed progressive oedema of the face and lower limbs, together with decreased urinary output and weight gain (approximately 10 kg).
On examination, the patient was calm and conscious without any respiratory, urinary or gastrointestinal symptoms. On physical examination she was afebrile, hypertensive (blood pressure 157/98 mmHg), tachycardic (101 beats per minute), had remarkable pitting oedema, dependent on gravity, with low oxy- gen saturations (88%) yet without dyspnoea. She had neither hepatosplenomegaly nor signs of cardiac failure, such as jugular vein engorgement or orthopnoea. She had no sequelae from her previous stroke. The remainder of the physical examination was normal. An indwelling urinary catheter was introduced for diuresis evaluation. Blood and urine cultures were obtained.
Her initial blood results revealed average haemoglobin level (12.5g/dL), normal platelet count (156 000/uL), normal serum creatinine (0.9 mg/dL), hypoalbuminaemia (1.9 g/dL) and hyperlipidemia (total cholesterol 372 mg/dL, high density lipoprotein 48 mg/dL, low density lipoprotein 215 mg/dL, triglycer- ides 207 mg/dL). Mid-stream urinalysis demonstrated proteinuria (++++) and microscopic haematuria (+/-) and the 24-hour urine collection estimated 16 g of proteinuria. Electrocardiogram was normal but chest radiograph revealed a bilateral pleural effusion suggestive of pulmonary congestion. Renal ultrasound scan showed normal sized kidneys with no signs of hydronephrosis.
Upon these findings, a diagnosis of NS was sug- gested and the patient was immediately started on intravenous diuretics (furosemide 80 mg 1id), anti-hypertensive drugs (enalapril 20 mg 1id, losartan 50 mg 1 id) and subcutaneous enoxaparin (1.5 mg/kg per 24 hours) on therapeutic dosage before she was transferred to the Nephrology Department.
On arrival, additional medical history was obtained and diagnostic procedures were continued. The patient had two almost complete gestations (twins) under medication with warfarin and no family history of vascular-associated disorders. She had been living in the countryside and worked as a housewife. She had no known allergies or history of previous infec- tions or recent medications.
Further investigation displayed negative antinu- clear antibodies (ANAs) with normal complement levels (C3, C4), negative anti-neutrophil cytoplasm antibodies (ANCA), negative cryogobulins, negative anti-glomerular basal membrane (anti-GBM), decreased IgG (467 mg/dL) with normal IgA (259 mg/ dL) and IgM (105 mg/dL) levels on immunofixation serum test, increased ß2 and 2 on electrophoresis, ß2 microglobulin and erythrocyte sedimentation rate (ESR). The ratio between serum and urine light chains was normal as well as antithrombin III. Antiphospholipid antibodies (anticardiolipin, anti-ß2-glyco-protein 1, lupus anticoagulant) were negative. Finally, serological virology for Hepatitis B, C and Human Immunodeficiency virus was negative as well.
In order to pursue a diagnosis, a percutaneous renal biopsy was performed (2 corticomedullary fragments were collected – one for light microscopy and other for immunofluorescence). Light microscopic evaluation (Figs. 1 and 2) showed no glomerular changes other than sclerosis as a result of ischaemia. Of the 10 glomeruli, only 1 was completely intact, 4 were partially intact and 5 were sclerosed. The first 5 intact glomeruli had no changes; however the remaining 5 were globally sclerosed as a result of ischaemia. Tubulointerstitium had areas of focal cortical atrophy (FCA) located in the subcapsular renal cortex. Capillaries had no thrombi inside but one was mildly occluded from intimal hyperplasia. Immunofluorescence (IF) microscopy was negative for glomerular deposits. Based on these findings and due to the scarce number of intact glomeruli (5), a primary podocytopathy (either MCD or FSGS) was thought to be the cause of NS and empirical treat- ment with corticosteroids (prednisolone 1mg/kg per day) was started; oral anticoagulation with warfarin was restored.
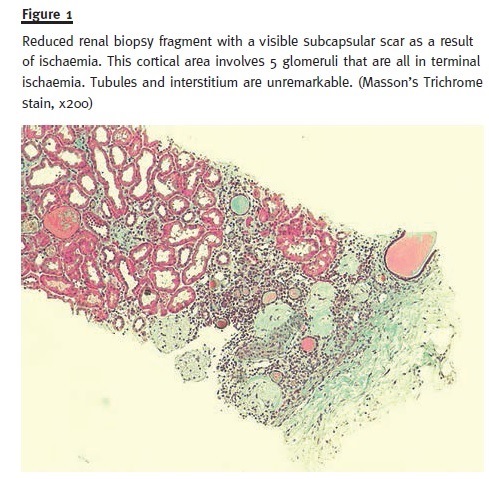
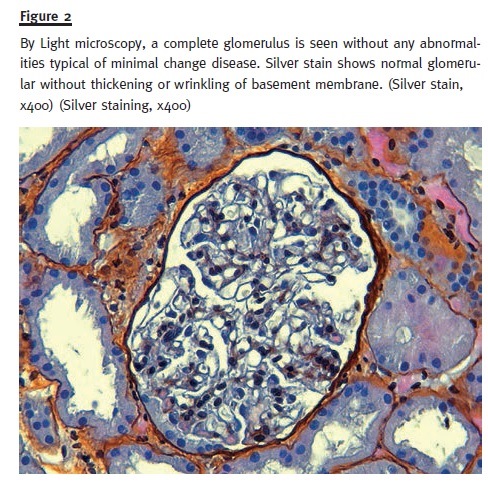
After 10 days, the patient showed a positive outcome with oedema regression and normal urinary output without the need for renal replacement therapy. Blood pressure control was achieved and pro- teinuria demonstrated a partial remission. She was discharged on corticosteroids therapy (1 mg/kg per day – 80 mg per day) and oral anticoagulation (war- farin 5 mg per day) until the next follow–up visit.
Two months from discharge, the patient returned to the outpatient care for a follow-up. She had complete regression of the oedema (with resultant weight loss 10 kg). Her serum biochemistry revealed an almost normalized serum albumin (3.4 g/dL) and lipid profile. The renal function remained stable and the 24-hour urine collection revealed a proteinuria of 52 mg. As the patient reached complete NS remission, two weeks after she was slowly tapered down on corticosteroids to 5 mg per day along 4 months. Nonetheless, by the end of that period, the patient relapsed with full-blown NS leading to a new cycle of prednisolone in higher doses (80 mg/ per day) for complete remission. Three months after, at the last follow-up, the patient was maintained on 10 mg of prednisolone every other day without significant proteinuria (< 10 mg/24 hours).
DISCUSSION
In this patient, the diagnosis of APS was based on her obstetrical and neurological complications, together with circulating APL, without renal manifestations. Ten years after she developed NS (with generalized gravity-driven oedema that developed over weeks, foamy urine and hypertension without renal dysfunction, severe hypoalbuminemia, dyslipidaemia and heavy proteinuria) without any clinical or serological manifestations of APS.
Antiphospholipid syndrome nephropathy (APSN) results from vascular-induced renal injury, which may range from microthrombosis of glomerular capillaries to thrombosis of the main renal artery and vein, with subcapsular renal cortical necrosis and infarction. Vascular lesions may be acute (resembling thrombotic microangiopathy) or chronic, associated with the development of vaso-occlusive process. Its renal manifestations may appear as asymptomatic haematuria, mild to nephrotic-range proteinuria, severe secondary renovascular hypertension or rapidly progressive renal failure2,6,7, which did not occur in our case. Lifelong anticoagulation to obtain an international normalized ratio (INR) ≥ 3.0 remains the mainstay treatment of patients with renal involvement due to APS2,5,6. Antiplatelet drugs, such as acetylsalicylic acid in combination with warfarin, may be effective as they act in synergism. Although immu- nosuppression and corticosteroids seem to reduce APL titres and inflammation, they do not decrease the risk of thrombotic events6, 8.
In our patient, the absence of clinical and biochemical findings characteristic of APSN, the negativity of the immunological study and the full blown NS was suggestive of a primary glomerulopathy superimposed in APS. Although quite rare, the association between primary glomerulopathies and primary APS, mainly membranous nephropathy but also MCD or pauci-immune glomerulonephritis, has been reported2,3,9. Yet, systemic diseases, such as SLE, have been described in patients with APS and APSN, to the best of our knowledge10.
As usual, renal biopsy was the key factor for the diagnosis of the underlying cause of NS. Opposed to what would be expected in a pure APS nephropathy2, 5-7, histological findings under light microscopy included an area of subcapsular focal cortical atrophy and mild intimal hyperplasia of only one capillary. No thrombotic microangiopathy with occluded ves- sels was seen. Half of the glomeruli seen showed no abnormalities and the other half were sclerotic as result of ischaemia. The glomerular immunofluorescence was negative. Unfortunately, electron microscopy was not performed due to technical problems.
Based on these findings, a primary podocyopathy (either characteristic of MCD or an initial phase of FSGS) was probably the cause of NS, superimposed in APS renal picture; the absence of electronic micros- copy and the paucity of intact (partially slcerosed) glomeruli precluded the immediate diagnosis of MCD11. However, the patient was started on warfarin and corticosteroids with an excellent and prompt response (proteinuria remission within 2 months)11-14. This fact, together with the sudden presentation of NS with heavy proteinuria and normal glomeruli at light microscopy favoured MCD over other glomerulopathies. The relapsing-remitting course of this NS (during steroid tapering, common in adult MCD) and the absence of progression to renal failure were two additional factors to corroborate the final diagnosis of MCD15.
Minimal change disease is usually corticosteroid-sensitive (1 mg/kg per day not exceeding 80 mg) and remission might take up to 15 weeks, mainly in adults15 On the contrary, even though at certain situations FSGS may respond positively to CTS it usually does not happen so frequently as in MCD. Few cases of MCD are steroid dependent (up to 30%) and 1⁄4 of patients are frequently relapsing, as described in our report13.
However, whether podocyte effacement is correlated with APL immunological mechanisms so many years after the diagnosis or is merely an unfortunate event, it is not known. In one hand, the significant amount of time that passed between these two clinical situations and the APL negativity during this episode did not show a probable correlation. Also, during this period (10 years after the initial diagnosis) the patient had no APL positive title, which makes it quite difficult to understand how they normalized without immunosuppressive treatment. Evidence suggests that patients with previous thrombotic manifestations have notably high APL and, yet, by the time of NS presentation this patient did not2. On the other hand, a possible association could result from an indolent interaction between autoantibodies (T-cell mediated) and the phospholipid components of renal podocytes leading to effacement and resultant defect in the glomerular filtration barrier to protein without APL positivity. However, even though there is lack of evidence that MCD may result from an antibody response, it is known that some autoimmune diseases (SLE) may be develop this glomerulopathy.
The purpose of this clinical case report was to highlight APS atypical renal involvement and to under-line the importance of looking beyond what we already know. A full background history, a serological profile, a high degree of suspicion and finally a renal biopsy were fundamental to reach the final diagnosis. Of note, an MCD diagnosis was determined on four main assumptions: sudden onset of NS, heavy pro- teinuria, absence of glomerular lesions on light microscopy (including immunoglobulin deposition) and complete remission of NS once CTS were started, despite unavailability of electron microscopy.
As far as we know, only a few cases have been reported with this association2,12. Even though, much has been elucidated concerning APS its pathogenesis and optimal treatment are still uncertain. It remains unclear whether MCD simply occurs concomitantly with APS or is linked to this syndrome by some common pathogenic pathway. Further research on APL immunological actions may be needed to elucidate these diseases in order to guide the treatment.
References
1. Giannakopoulos B, Krilis SA. The pathogenesis of antiphospholipid syndrome. N Engl J Med 2013;368(11):1033-1044. [ Links ]
2. Sciascia S, Cuadrado MJ, Khamashta M, Rocatello D. Renal involvement in antiphospholipid syndrome. Nat Rev Nephrol 2014;10(5):279-289. [ Links ]
3. Appel GB. Antiphospholipid syndrome and the kidney [internet]. [Place unknown]: UpToDate; 2014 [updated Sep 2014, cited Sep 03, 2014]. Available at: www.uptodate.com/contents/antiphospholipid-syndrome-and-the-kidney. [ Links ]
4. Bermas BL, Erkan D, Schur P. Diagnosis of the antiphospholipid syndrome [internet]. [Place unknown]: UpToDate; 2014 [updated Sep 2014, cited Sep 17, 2014]. Available at: http://www.uptodate.com/contents/diagnosis-of-the-antiphospholipid-syndrome. [ Links ]
5. Glassock RJ. Other Glomerular Disorders and the Antiphospholipid Syndrome. Comprehensive Clinical Nephrology. Philadelphia: Saunders Elsevier; 2015. p. 330-338. [ Links ]
6. Alchi B, Griffiths M, Jayne D. What nephrologists need to know about antiphospho-lipid syndrome. Nephrol Dial Transplant 2010;25(10):3147-3154. [ Links ]
7.Caraba A, Crisan V, Munteanu A, Serban C, Nicoara D, Romosan I. The Kidney in Antiphospholipid Syndrome, In: Antiphospholipid Syndrome, Bulikova A. (Ed). InTech, April 2012 [cited April 2012]. Available at: http://www.intechopen.com/books/antiphos-pholipid-syndrome/antiphospholipid-syndrome-and-the-kidney. [ Links ]
8. Butani L. End-stage renal disease from glomerulonephritis associated with antiphospholipid syndrome. Pediatr Nephrol 2004;19(7):812-814. [ Links ]
9. Fakhouri F, Noël LH, Zuber J, et al. The expanding spectrum of renal diseases associated with antiphospholipid syndrome. J Kidney Dis 2003;41(6):1205-1211. [ Links ]
10. Nzerue CM, Hewan-Lowe K, Pierangeli S, Nigel Harris EN. Black swan in the kidney: renal involvement in the antiphospholipid antibody syndrome. Kidney Int, 2002;62(3):733-744. [ Links ]
11. Mason PD, Hoyer PF. Minimal change nephrotic syndrome. In: Comprehensive Clinical Nephrology. Philadelphia: Saunders Elsevier 2015;208-217. [ Links ]
12. Rossiñol T, Cervera R, Lopez C, Solé M, Ramos-Casals M, Font J. Antiphospholipid syndrome and minimal change nephropathy. Letter to the editor. Lupus 2006;15(8):547- 548. [ Links ]
13. Fervenza FC, Sethi S. Frequent-relapsing, steroid-dependent minimal change disease: is rituximab the answer? Neprhol Dial Transplant 2014;29(4):722-727. [ Links ]
14. Joseph RE, Radhakrishnan J, Appel GB. Antiphospholipid antibody syndrome and renal disease. Curr Opin Nephrol Hypertens 2001;10(2):175-181. [ Links ]
15. Nachman PH, Jennette C, Falk RJ. Primary glomerular disease. In: Brenner BM. Brenner & Rectors The Kidney. 8th ed. Philadelphia: Elsevier Saunders; 2012:1100-1168. [ Links ]
Dra Alice Lança
Department of Nephrology
Centro Hospitalar Médio Tejo, Hospital de Torres Novas
Av. Xanana Gusmão s/n 2350-754 Torres Novas, Portugal.
E-mail: alicelancabaptista@gmail.com
ACKNOWLEDGMENTS
The authors would like to take this as an opportunity to thank Dr Maria Fernanda Carvalho (Kidney Morphology Laboratory, Department of Nephrology, Hospital Curry Cabral, Lisbon, Portugal) for her avail- ability and dedication in sharing and discussing the histopathological renal biopsy specimens with us.
Conflict of interest statement: None declared.
Received for publication: 05/08/2015
Accepted in revised form: 04/09/2015

