










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.30 no.1 Lisboa mar. 2016
CASE REPORT
Bartter syndrome – report of an unusual late presentation case and brief review
Síndrome de Bartter – um caso raro de apresentação tardia, e breve revisão
Bernardo M Costa, Joaquim Calado, David Navarro, Fernando Nolasco
Department of Nephrology, Renal Transplantation Unit, Hospital Garcia de Orta, Almada, Portugal.
ABSTRACT
Bartter syndrome is a rare autosomal recessive condition caused by the inability of the thick ascending limb to reabsorb filtered sodium and chloride. Types I and II, called antenatal Bartter syndrome, are the most severe, and manifest in-utero as polyhydramnios, preterm labour, salt wasting, life-threatening volume depletion, and severe hypokalemic metabolic alkalosis, with a high early mortality rate if untreated. Type III is called classic Bartter syndrome and is usually milder and often diagnosed later, in early adolescence. Type IV is as severe as types I and II, but courses with sensorineural deafness. Type V is the latest entry to the Bartter-like syndromes. The defective transporter proteins responsible for these subtypes have been identified and their mutations have been characterized using genetic sequencing and in vitro heterologous expression models. We present an unusual case of a very late diagnosis of an attenuated type IV Bartter syndrome. Our suspicion of the G47R mutation in the ß-subunit (Barttin) of the ClC-K chloride channels was confirmed by genetic sequencing. This is a second unrelated case in our centre. Although there is significant variability in the presentation of some subtypes of Bartter syndrome, there is still a strong genotype-phenotype correlation in some mutations, like the one we present here. The acknowledgement of this has provided insight into the genetic and molecular mechanisms of the disease.
Key-Words: Bartter syndrome; Bartter syndrome, type 4A; BSND protein, human; hypokalemia; mutation; renal tubular transport, inborn errors.
RESUMO
O síndrome de Bartter é uma doença autossómica recessiva rara, causada pela incapacidade da segmento ascendente da ansa de Henle de reabsorver o cloro e sódio filtrados. Os tipos I e II, chamados síndrome de Bartter pré-natal, são os mais graves. Manifestam-se in-utero por polihidrâmnios, parto prematuro, perda urinária de sal, contração de volume, e hipocaliémia grave com alcalose metabólica, apresentando uma elevada mortalidade precoce se não tratado. O tipo III é chamado síndrome de Bartter clássico, tipicamente é menos grave e só é diagnosticado frequentemente na adolescência. O tipo IV é tão grave como os tipos I e II, mas é acompanhado de surdez neurosensitiva. O tipo V foi o último a juntar-se aos síndromes Bartter-like. As proteínas transportadoras não-funcionantes responsáveis pelos vários subtipos já foram identificadas e as suas mutações caracterizadas, graças à sequenciação genética, e a modelos de expressão heteróloga in vitro.
Apresentamos um caso raro de diagnóstico tardio de Síndrome de Bartter tipo IV. A suspeita de estarmos perante a mutação G47R na subunidade ß (Barttina) dos canais de cloro ClC-K foi confirmada por sequenciação genética. Este é o segundo caso no nosso centro (não-aparentados). Apesar de haver uma variabilidade significativa na apresentação de alguns subtipos de síndrome de Bartter, existem fortes correlações genotipofenotipo em algumas mutações, como a que apresentamos aqui. A constatação deste facto tem proporcionado o aumento do nosso conhecimento sobre os mecanismos genéticos e moleculares desta doença.
Palavras-Chave: Erros congénitos; mutação; hipocaliémia; proteína BSND, humana; síndrome de Bartter; síndrome de Bartter, tipo A4; transporte nos túbulos renais.
INTRODUCTION
The healthy glomerulus filters large volumes of blood through a barrier that is semipermeable to molecules, depending on their size and electric charge. Very small molecules like water and electrolytes are filtered easily and enter the renal tubules in massive amounts that must be reabsorbed in order to maintain homeostasis. Bartter syndrome (BS), first described in 1962, is one of the hereditary salt-wasting tubulopathies which have been increasingly better characterized over the past 20 years through the identification of the malfunctioning transmembrane transporters and the mutations responsible for those defects1.
Bartter syndrome is a rare condition caused by the inability of the thick ascending limb (TAL) to actively reabsorb filtered sodium and chloride. This can be caused by the malfunctioning of any of four interdependent gene products (Fig. 1), namely: 1) the apical furosemide-sensitive Na-2Cl-K cotransporter (NKCC2) encoded by the SLC12A1 gene2,3; 2) the apical potassium ROMK channel (KCNJ1 gene)4; 3) the basolateral chloride channel ClC-Kb (CLCNKB gene)5; and 4) the chloride channels ß-subunit Barttin (BSDN gene) necessary for the activation of both ClC-Kb and ClC-Ka6. Inactivating mutations in any of these genes produce the different subtypes of Bartter syndrome (types I, II, III, and IV respectively), which are all autosomal recessive. Additionally, other molecules, like the basolateral calcium-sensing receptor (CASR) and the intracellular signalling protein WNK can modulate the expression of these transporters, and produce Bartter-like syndromes7. The constitutional activation of CASR inhibits the activity of NKCC2 and is now called BS type V8.
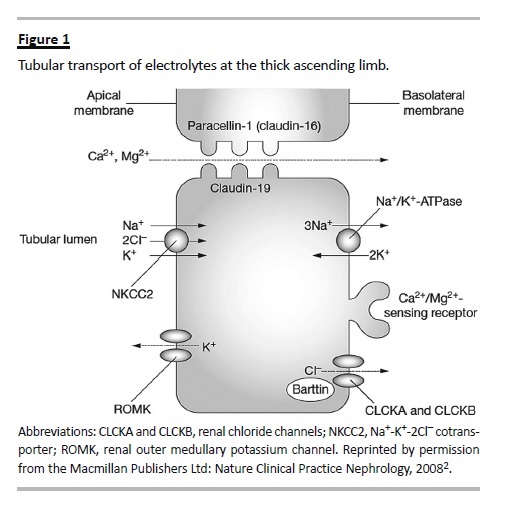
Other conditions that induce extra-renal chloride and volume losses with similar metabolic abnormalities are sometimes called Pseudo-Bartter syndromes.
Examples of this are cystic fibrosis, abuse of laxatives and bulimia, all of which can be easily ruled out with a low urinary chloride measurement. Surreptitious diuretic use, on the other hand, can only be ruled out by careful interrogation of the patient or a positive urinary drug test9.
Phenotypically, the four subtypes of Bartter syndrome manifest differently. Types I and II constitute what is termed antenatal Bartter syndrome (sometimes called hyperprostaglandin E2 syndrome, because high urinary prostaglandin levels were used for its diagnosis). They are the most severe. They manifest first in-utero with polyhydramnios, and infants exhibit polyuric hypostenuria, life-threatening volume depletion, low blood pressure, impaired development and high early mortality if left untreated. Some, but not all, develop renal failure. Types I and II also exhibit hypercalciuria and nephrocalcinosis, since the paracellular reabsorption of calcium and magnesium depends on the secretion of potassium to the lumen, in order to create a favourable electrochemical gradient10.
Type III is termed classical Bartter syndrome. Usually it is clinically milder (due to partial compensation by the functioning ClC-Ka chloride channel), manifests in early adolescence, and may mimic Gitelman syndrome, including its characteristic hypomagnesaemia and hypocalciuria11. It should be noted though that Bartter syndrome type III is highly variable, and severe cases have been reported. Type IV is called antenatal Bartter syndrome with sensorineural deafness. Its presentation is similar to that of types I and II, with the addition of a hearing deficit. This is because both chloride channels are expressed and have a functional role at the inner ear epithelium12.
Common to all types of BS, patients exhibit hyperreninemic hyperaldosteronism secondary to volume depletion. This induces a persistent hypokalaemic metabolic alkalosis due to the distal tubule effects of aldosterone, which increases distal sodium absorption at the expense of increased K+ and H+ secretion.Another common finding is overproduction of prostaglandin E2, as a consequence of salt wasting, which further reduces sodium chloride absorption at the TAL. This is the reason why inhibitors of cyclooxygenase like indomethacin have long been used in the treatment of severe cases.
Although there is significant variability in the presentation of some subtypes of BS, there is still a strong genotype-phenotype correlation in some mutations, like the one we present in the following case.
CASE REPORT
We present a patient first referred to us at the age of 59. His parents were 2nd degree cousins. The gestation period was unremarkable aside from mild polyhydramnios.
He was congenitally deaf, had minor cognitive and motor impairment, epilepsy and bilateral inguinal hernias. He was referred to us for the evaluation of persistent hypokalaemia and low blood pressure, which were being treated with oral KCl supplementation (24 mEq/day) and etilefrine. Relevant serum and urinary biochemical data are summarized in (Table I). Blood tests showed hyperreninaemic hyperaldosteronism with metabolic alkalosis, hypokalaemia and hypochloremia.
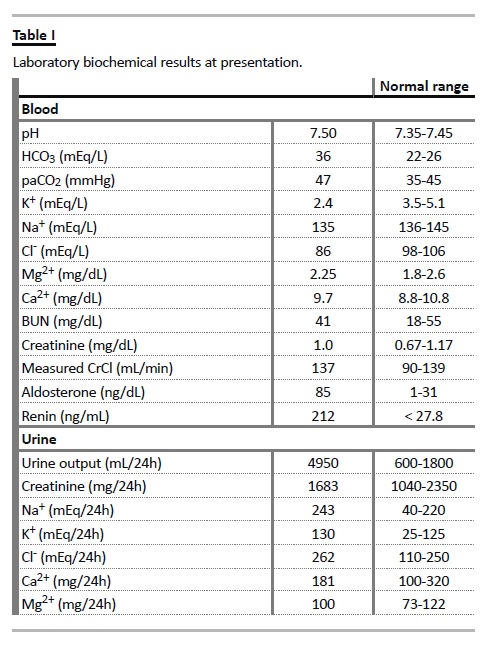
Serum magnesium level was normal. The 24h urine collection showed hypernatriuria, hyperkaliuria, and hyperchloriuria.
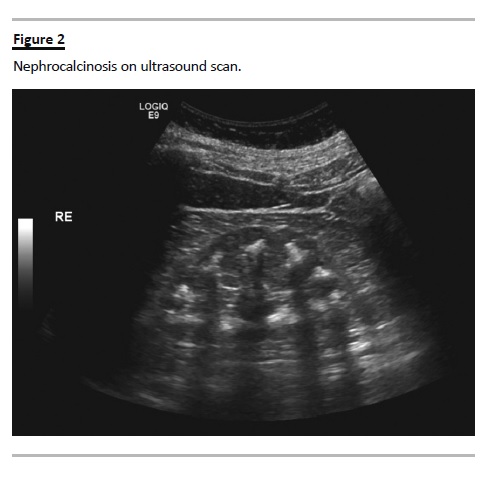
Urinary calcium levels were normal. BUN, creatinine and measured creatinine clearance were normal. Nephrocalcinosis was evident in the ultrasound and plain abdominal radiograph (Fig. 2). In spite of the patients age, the metabolic derangements and hearing impairment led us to consider Bartter syndrome type IV. Genetic testing confirmed homozygotic substitution of c.139G>A in the BSND gene resulting in Gly to Arg at position 47 (p.G47R) (Fig. 3).

The oral KCl supplementation was increased to 72 mEq/day, with significant improvement of the hypokalaemia (3.3 mEq/L; reference 3.5-5.0 mEq/L) and no further therapeutic interventions were considered necessary.
DISCUSSION
From our review of the literature, we have identified 10 published cases of late onset attenuated Bartter syndrome type IV due to the G47R mutation, 1 of them from our centre13-16. The patient we present here is unrelated to the previous one and, to the best of our knowledge, this is the oldest patient at the time of diagnosis.
Most of the clinical features and biochemical results presented in this case are in accordance with the previously published cases. The most intriguing aspect of this case is the absence of hypercalciuria, given the presence of frank nephrocalcinosis. This has already been described in a patient with unexplained reduced glomerular filtration rate, and it was speculated that as the renal insufficiency progressed, the lower filtered calcium load would be effectively handled by alternative calcium channels, and any hypercalciuria that might have been present initially, would afterwards subside15.
In our past case, glomerular filtration was normal, and hypercalciuria was persistent, which is compatible with this hypothesis. Conversely, in the present case we did not find hypercalciuria even though the creatinine clearance was normal. This suggests that either other mechanisms that account for the resolution of the hypercalciuria are in place, or that nephrocalcinosis is dependent on other factors besides the increased calcium loss. A possible example of this is hypocitraturia. Garcia-Nieto et al. found hypocitraturia in 4 out of the 5 cases of G47R variant of BS-IV they reported on, including one with nephrocalcinosis, normal creatinine clearance and no hypercalciuria13. Unfortunately, due to the small number of reported cases, the other factors involved in the calcification process in this rare phenotype have not been characterized.
Another interesting aspect of this case is the good response to oral KCl supplementation alone. The treatment of more severe Bartter syndrome cases usually requires the addition of a non-steroidal anti-inflammatory drug (NSAID) (in order to decrease prostaglandin production and reduce the filtration rate, water, and salt wasting) and sometimes a potassium sparing diuretic (spironolactone or amiloride). However, these medications carry a risk of chronic renal toxicity, gastrointestinal and cardiovascular adverse events, volume depletion and severe electrolyte disturbances11. Since the G47R variant of BS-IV allows for some residual function of ClC-K channels, it is probable that the potassium wasting is less severe and, hence, the response to supplementation is better than that of more severe phenotypes. In our small experience (2 cases), potassium supplementation was enough.
Once more, due to the small number of cases and unreported results of the therapeutic interventions there is not enough data to make confident statements about the treatment of this variant.
We believe that these cases are relevant because they allow us to create strong clinical-to-molecular correlations.
When this BSND mutation and its unusual presentation were first identified, it was speculated that it might originate a protein that retained some residual function, which would explain its milder phenotype. Using confocal microscopy and an in vitro model with heterologous expression of disease-causing mutations of the BSND gene in MCKD cells, Jansen GH et al. showed how three missense mutations and two nonsense mutations originate non-functioning proteins. This was because the mutated Barttin protein would either not allow the migration and insertion of the channel in the basolateral membrane, or its activation, or both. Another missense mutation that they studied was the G47R, which produces a mutant Barttin that binds weakly to ClC-K chloride channels, leaving large amounts of the unbound channel wandering in the cytoplasm, and significantly reducing the chloride conductance of the cells17.
Nevertheless, the same mutation may originate significantly different phenotypes, which is especially evident in BS type III. It has been speculated that this may be because of individual variability in the genes of additional channels and transporters (like the ClC-Ka, the NaCl cotransporter, or the CFTR), or because of variability in modifying genes that regulate expression18. Interestingly, one case of antenatal Bartter syndrome with sensorineural deafness without a mutated BSND gene has been reported, where the researchers cleverly deduced and subsequently demonstrated that both ClC-Kb and ClC-Ka had simultaneous inactivating mutations19. This has been classified as BS type IVB, as opposed to BS type IVA, which is due to mutations in Barttin.
In conclusion, Bartter syndrome is a fascinating genetic disorder that has greatly improved our understanding on the mechanisms of renal tubular solute transport and its genetic foundations, which despite our great advances, continues to pose a diagnostic challenge both at the consultation room and at the laboratory.
References
1. Bartter FC, Pronove P, Gill JR, MacCardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med 1962; 33:811–828. [ Links ]
2. Naderi AS, Reilly RF Jr. Hereditary etiologies of hypomagnesemia. Nat Clin Pract Nephrol 2008;4(2):80-89. [ Links ]
3. Simon DB1, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartters syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 1996 Jun;13(2):183-188. [ Links ]
4. Simon DB, Karet FE, Rodriguez-Soriano J, et al. Genetic heterogeneity of Bartters syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 1996;14(2):152-156. [ Links ]
5. Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartters syndrome type III. Nat Genet 1997;17(2):171-178. [ Links ]
6. Birkenhäger R, Otto E, Schürmann MJ, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 2001; 29(3):310–314. [ Links ]
7. Kleta R, Bockenhauer D. Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol 2006;104(2):73-80. [ Links ]
8. Vargas-Poussou R, Huang C, Hulin P, et al. Functional characterization of a calciumsensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol 2002;13(9):2259-2266. [ Links ]
9. Amirlak I, Dawson KP. Bartter syndrome: an overview. QJM 2000; 93(4):207–215. [ Links ]
10. Kurtz I. Molecular pathogenesis of Bartter and Gitelmans syndromes. Kidney Int 1998;54(4):1396-1410. [ Links ]
11. Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol 2008;4(10):560-567. [ Links ]
12. Estévez R, Boettger T, Stein V, et al. Barttin is a Cl-channel beta-subunit crucial for renal Cl-reabsorption and inner ear K+ secretion. Nature 2001; 414(6863)558-561. [ Links ]
13. Garcia-Nieto V, Flores C, Luis-Yanes MI, Gallego E, Villar J, Claverie-Martín F. Mutation G47R in the BSND gene causes Bartter syndrome with deafness in two Spanish families. Pediatr Nephrol 2006; 21(5):643–648. [ Links ]
14. Brum S, Rueff J, Santos JR, Calado J. Unusual adult-onset manifestation of an attenuated Bartter syndrome type IV renal phenotype caused by a mutation in BSND. Nephrol Dial Transplant 2007;22(1): 288-289. [ Links ]
15. Miyamura N, Matsumoto K, Taguchi T, et al. Atypical Bartter syndrome with sensorineural deafness with G47R mutation of the beta-subunit for ClC-Ka and ClC-Kb chloride channels, barttin. J Clin Endocrinol Metab 2003;88(2):781-786. [ Links ]
16. Park CW, Lim JH, Youn DY, et al. Renal dysfunction and barttin expression in Bartter syndrome Type IV associated with a G47R mutation in BSND in a family. Clin Nephrol 2011;75 (Suppl 1):69-74. [ Links ]
17. Janssen AG, Scholl U, Domeyer C, Nothmann D, Leinenweber A, Fahlke C. Diseasecausing dysfunctions of barttin in Bartter syndrome type IV. Am Soc Nephrol 2009;20(1): 145–153. [ Links ]
18. Konrad M, Vollmer M, Lemmink HH, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol 2000;11(8):1449-1459. [ Links ]
19. Schlingmann KP, Konrad M, Jeck N, et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 2004;350(13):1314-1319. [ Links ]
Dr. Bernardo Costa
Department of Nephrology, Hospital Curry Cabral – Centro Hospitalar Lisboa Central, EPE,
Rua da Beneficência, nº 8, 1069-166 Lisbon, Portugal.
E-mail: bmarquesdacosta@gmail.com
Disclosure of Potential Conflicts of Interest: None declared.
Received for publication: 27/09/2015
Accepted in revised form: 11/11/2015

