










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.30 no.2 Lisboa jun. 2016
MINI-REVIEW
Calcific uraemic arteriolopathy – A mini-review
Filipa Brito Mendes1, Sofia Couto Rocha2, Rodica Agapii2, Ana Silva1,3, André Fragoso1, Teresa Jerónimo1, Ana Pimentel1, Pedro L Neves1,3
1 Department of Nephrology, Algarve Hospital Centre, Faro, Portugal
2 Department of Internal Medicine, Algarve Hospital Centre, Portugal
3 Department of Biomedical Sciences and Medicine, Faro, Portugal
ABSTRACT
Calcific Uraemic Arteriolopathy (CUA) or calciphylaxis, is a thrombotic disorder of skin and subcutaneous tissue which typically presents with painful purpuric nodules that may progress to necrotic ulcers, and is a severe, life‑threatening condition. CUA is an uncommon clinical entity that affects mostly haemodialysis (HD) patients.
Although the process of vascular calcification was initially thought to be the result of a passive deposition of calcium‑phosphate crystals, current knowledge suggests a distinct mechanism, including cellular activity with differentiation of vascular smooth muscle cells (VSMCs) into chondrocyte as well as osteoblast‑like cellular phenotypes and deficiencies in calcification inhibitors. Although multiple studies suggest a potential relationship between warfarin and CUA, larger prospective studies are needed in order to better evaluate this association, and randomised controlled trials are needed to assess the benefit of distinct interventions in this setting. In this article the topic of CUA is reviewed based on a clinical case of a 65‑year‑old man undergoing haemodialysis, who underwent an aortic valve replacement one year earlier, receiving a mechanical heart valve, and who has been under warfarin therapy since then.
Key‑Words: Calciphylaxis, Fetuin‑A, Gas‑6, MGP, Vitamin K, Warfarin.
INTRODUCTION
Calcific Uraemic Arteriolopathy (CUA), also known as calciphylaxis, is a severely morbid and life‑threatening condition. It is characterised by medial vascular calcification, affecting small arterioles, and results in ischaemic subcutaneous necrosis with vulnerable skin ulcerations1.
Despite being rare, it is increasingly identified and reported worldwide2.
CUA usually occurs in patients with chronic kidney disease (CKD), mostly in advanced stages of kidney disease (ESRD).
We report a case of calciphylaxis occurring after starting warfarin therapy.
CASE SUMMARY
We report the case of a 65‑year‑old male gender patient with end‑stage renal disease (ESRD) due to diabetes mellitus type 2, who had been on haemodialysis for the last 6 years. The patient had a body mass index of 25 Kg/m2, and had recently switched to haemodiafiltration with a dialysate composition of 1.50 mmol/L of calcium, 0.50 mmol/L of magnesium and 32 mmol/L of bicarbonate. The patient underwent an aortic valve replacement one year earlier, receiving a mechanical heart valve, and he has been under warfarin therapy since then. Secondary hyperparathyroidism was not a major problem, as the patient maintained serum parathormone levels in the range of 200 to 400 pg/mL for the last few years (Table I) taking sevelamer 1600mg/day and cinacalcet 90mg/week.
On February 2014, the patient was referred to the Nephrology department due to a painful violaceous lesion of 3 cm diameter on the dorsal region of the right leg.
A diagnosis of CUA was considered. The frequency of haemodialysis sessions was increased to 5 per week (4 hours x 5 /week); the dialysates calcium was reduced to 1.25 mmol/L, and warfarin was stopped and replaced by low molecular weight heparin (1mg/Kg/day). Sodium thiosulfate (STS) therapy was initiated the first day of hospital stay, which was one week after the onset of the clinical picture. The patient received STS treatment 3 x /week at the end of dialysis session, at a 25g dose with no side‑effects.
Mineral metabolism parameters (Table 1), calcium, phosphorus and parathormone levels were within acceptable limits. Hand and foot X‑rays confirmed ectopic calcifications (Fig. 1). Blood cultures were negative and arterial and venous eco‑doppler of lower limbs was normal.
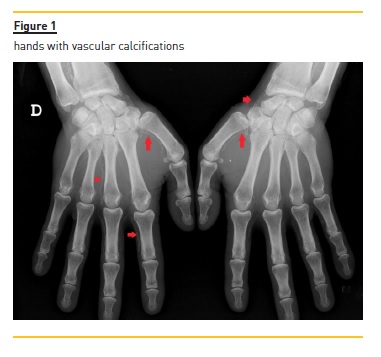
On the fourth day of hospital stay, a skin biopsy was performed and showed epidermis atrophy with focal necrosis as well as vascular calcifications in deep dermis.
The biopsy, performed in the smallest lesion, had a proper healing. However, despite all therapeutic measures, the skin lesion continued to increase in dimension while numerous other identical skin lesions appeared in the region.
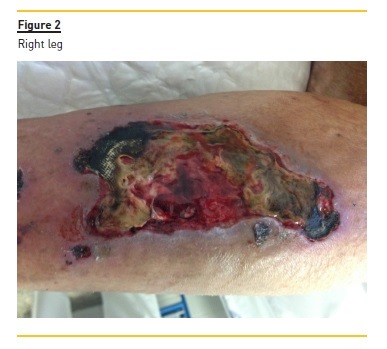
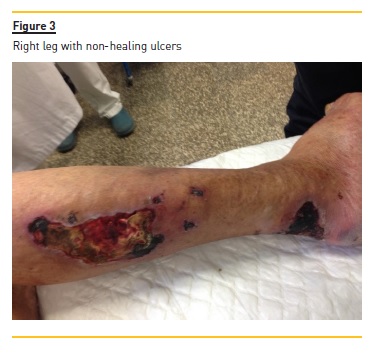
The confluence of these small isolated lesions, which subsequently suffered necrosis and non‑healing ulcerations (Fig. 2 and 3), led, after three months, to bilateral above‑knee amputation. By that time, STS was stopped.
At two years of follow‑up, the patient did not show any additional complications and was alive and on chronic haemodialysis.
MINI‑REVIEW
CUA is estimated to occur in 1% of patients with chronic renal failure and 4% of patients undergoing haemodialysis2-5 and is associated with a high morbidity and a mortality rate of 60%‑80%, most commonly caused by infectious complications, culminating in sepsis and organ failure2,3.
Several risk factors for CUA have been suggested, such as female gender, diabetes mellitus, obesity, hyperphosphataemia, ESRD with severe secondary hyperparathyroidism and the use of warfarin or of calcium‑based phosphate binders2,3.
The insights into the pathogenesis of vascular calcification in CKD have changed significantly. Until recently, the process of vascular calcification was thought to be merely the result of a passive deposition of calcium‑phosphate crystal2,6. However, current evidence suggests it is an active and coordinated process which not only involves cellular activity with differentiation of vascular smooth muscle cells (VSMCs) into chondrocytes, but that it also encompasses osteoblast‑like cellular phenotypes and deficiencies in calcification inhibitors.
This process is initiated by the uraemic milieu, which is characterised by the presence of hyperphosphataemia, uraemic toxins and reactive oxygen species, although it is also associated with a decrease in vascular calcification inhibitor proteins3,4.
Fetuin A/α2‑Heremans Schmid glycoprotein (AHSG), a 48‑kDa protein, acts as a negative regulator of soft‑tissue calcification, and it is synthetised in the liver and then secreted into circulation. AHSG acts as a systemic inhibitor of hydroxyapatite synthesis, and is reduced in states of renal failure, inflammation and patients with CUA2,4,7,8.
Some other important inhibitors of vascular calcification are Matrix Gla protein (MGP) and Growth arrest‑specific gene 6 (Gas‑6), assuming special importance in patients taking vitamin K antagonists, as both are vitamin K dependent proteins (VKDPs)9.
MGP, which was first described in 1983 and is an 84‑amino acid protein produced by bone and VSMCs, acts as a local inhibitor of calcification4,5,10. It binds to insoluble calcium salts preventing hydroxyapatite crystals growth and, by blocking osteo‑inductive properties of bone morphogenic protein‑2, inhibits VSMCs from differentiating into osteoblast‑ like phenotypes3,5,6.
Although studies by Murshed et al. have demonstrated that MGP is also produced by the liver, such hepatic production does not seem to be protective against vascular calcification5,8. In CKD patients, MGP function appears to be impaired9.
Gas‑6 plays a double role. On the one hand, Gas‑6, produced by platelets, promotes thrombus formation as it contributes to platelet degranulation and aggregation, and on the other hand, Gas‑6 produced by VSMCs, by binding the receptor Axl, stimulates the anti‑apoptotic protein Bcl‑2 and inhibits the proapoptotic protein caspase 3, which allows it to play a protective role against vascular injury.5 The exact role of Gas‑6 and MGP in humans still needs further confirmation studies.
Both Gas‑6 and MGP share a common characteristic – they are VKDPs as they need to undergo gamma‑carboxylation, a vitamin K dependent process, in order to achieve full biological activity3,4,9.
Warfarin shares a common ring structure with vitamin K, interfering with vitamin K epoxide reductase (VKOR), an enzyme responsible for the reduction of the naturally oxidised form of vitamin K, which interrupts the recycling of this vitamin and ultimately the necessary gamma‑carboxylation of VKDPs5,8. There are two subtypes of vitamin K, vitamin K1, a plant‑synthesised phylloquinone, needed for hepatic carboxylation of coagulation factors which activate them and vitamin K2, a family of menaquinones which are important for the peripheral gamma‑carboxylation of proteins such as MGP and Gas‑6 produced and locally activated at VSMCs7. Animal and clinical studies support the concept that hepatic and peripheral carboxylation have unique vitamin K dependence. Vitamin K2 mainly affects peripheral carboxylation so that supplementation prevents arterial calcification while vitamin K1 mostly affects hepatic carboxylation and its supplementation does not prevent vascular calcification. Phylloquinone (vitamin K1) is found in green leafy vegetables. The exact source of vitamin K2 is controversial. Some authors claim that it comes either from enterocyte conversion of vitamin K1 or from indigenous intestinal bacteria5. Others state fermented food, such as cheese, is the main source of menaquinones6. Despite that, both vitamins K1 and K2 are absorbed from the small ileum and jejunum and both are affected by warfarin use10.
Hepatic carboxylation for coagulation factors activation has priority when compared to peripheral carboxylation of VKDPs. So, the first sign of a low functional vitamin K status is the incomplete carboxylation of extrahepatic Gla proteins, resulting in desphospho‑uncarboxylated matrix Gla protein (dp‑ucMGP) increase. The dp‑ucMGP is inversely correlated with the amount/function of vitamin K and its levels are significantly higher in patients under VKA, among other situations. Only further studies may demonstrate its value as a risk marker for cardiovascular disease and mortality8. According to recent data, low doses of warfarin are able to inhibit peripheral carboxylation, while they have no effect on hepatic carboxylation5.
Some studies with supplementation of vitamin K have been conducted. Spronk et al. found in rodents that menaquione‑4 inhibited warfarin‑induced arterial calcification, while phylloquinone did not8. In human studies it has also been shown that menaquinone supplementation decreased dp‑ucMGP plasma levels6,8,9.
Many researchers suggest that warfarin usage may be a risk factor for the development of CUA by the inhibition of VKDPs. Price et al. have shown that warfarin, in doses which inhibit gamma‑carboxylation of MGP, induces widespread vascular calcification in rodents and that its combination with Vitamin D–induced hyperphosphataemia exacerbates vascular calcification in young growing rats and not in mature ones. Those calcifications occur more often in medium‑sized vessels, sparing capillaries and veins, and predominantly involve the elastic lamellae of the media, sparing the intima. It was also reported, by Hayashi et al, that warfarin increased the risk of calciphylaxis 10‑fold3. Booth et al. have shown that subclinical vitamin K deficiency (normal coagulation studies but low concentrations of vitamin K1) is present in almost 30% of dialysis patients, combined with elevated serum phosphate levels. Patients with end‑stage renal disease show an increased risk of vascular calcification associated with warfarin use as compared with that of the general population, which might be explained by the use of vitamin D and diminished MGP levels observed in the ESRD population5.
However, these data on the role of Gas‑6 and MGP have not been confirmed in human studies 5.
The registry of the German section of the International Collaborative Calciphylaxis Network (ICCN) has shown that about half of all calciphylaxis patients have been treated with vitamin K antagonists before the onset of symptoms. Furthermore, a Japanese survey among dialysis centres estimated the risk of calciphylaxis to be 11‑fold higher in patients under warfarin as compared with those patients not under VKA treatment9. However, these results may be biased by confounding by indication due to the observational nature of these studies.
Some data suggest that some patients may show an increased susceptibility to warfarin, due to genetic polymorphism in carboxylation enzymes which leads to a wide range of VKDP phenotypes, something that may explain why many people under this drug do not develop CUA5.
CUA typically presents as firm painful purpuric plaques and nodules, surrounded by erythema and livedo reticularis. Skin lesions may progress to soft‑tissue ulceration, necrosis and non‑healing ulcerations2,3.
The lower extremities are involved in 90% of the cases and are the most frequent site involved, but skin lesions can occur in multiple other sites3.
Skin biopsy usually shows medial calcification and intimal hyperplasia of small and medium‑sized arteries, primarily in dermal and subcutaneous tissues. Inflammation with endovascular fibrosis and microthrombi are usually also present2-4. Nevertheless, the benefit of performing a skin biopsy should be weighed against the risk of profuse bleeding which is sometimes associated with a poor healing in CUA.
Several case reports and small case series have suggested that STS, a cations chelator, may be useful in treating calciphylaxis. However, published data has shown inconsistent results2,3,11.
Nigwekar et al. studied 172 patients from North America undergoing maintenance haemodialysis who developed CUA and were treated with STS. They observed a clinical improvement in the majority of patients as well as a lower 1‑year mortality compared with historical data from patients not treated with STS12.
However, there is no effective treatment to treat this disorder. Suggested treatment usually includes several distinct measures such as initiation of non‑calcium‑based phosphate binder, discontinuation of calcium‑based phosphate binder, initiation of cinacalcet, discontinuation of vitamin D compounds, lowering of dialysate calcium, discontinuation of warfarin, increased frequency of haemodialysis sessions, surgical parathyroidectomy, and wound care 12 However, there is no proof of benefit of any of these measures.
Randomised controlled trials are needed to better understand whether warfarin increases the risk for CUA, as well as whether vitamin K2 supplementation and other interventions could be therapeutic options in order to improve the management of this devastating condition. Until that data is available from human trials, no definitive answers can be provided on the role of warfarin in CUA or on the best treatment for CUA.
References
1. Hayden MR, Tyagi SC, Kolb L, Sowers JR, Khanna R. Vascular ossification‑calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis‑calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol 2005, 4:4. [ Links ]
2. Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev 2010, 3: 109–21. [ Links ]
3. Saifan C, Saad M. Warfarin‑induced calciphylaxis: a case report and review of literature. Int J Gen Med 2013, 6:665. [ Links ]
4. Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: A systematic review. Clin J Am Soc Nephrol. 2008, 3:1139–43. [ Links ]
5. Danziger J. Vitamin K‑dependent proteins, warfarin, and vascular calcification. Clin J Am Soc Nephrol. 2008, 3:1504–10. [ Links ]
6. Caluwé R, Vandecasteele S, Van Vlem B, Vermeer C, De Vriese AS. Vitamin K2 supplementation in haemodialysis patients: a randomized dose‑finding study. Nephrol Dial Transplant 2014, 29:1385–90. [ Links ]
7. Cadavid JC, DiVietro ML, Torres EA, Fumo P, Eiger G. Warfarin‑induced pulmonary metastatic calcification and calciphylaxis in a patient with end‑stage renal disease. Chest. 2011, 139:1503–6. [ Links ]
8. Theuwissen E, Smit E, Vermeer C. The role of vitamin K in soft‑tissue calcification. Adv Nutr 2012, 3:166–73. [ Links ]
9. Ketteler M, Rothe H, Brandenburg VM, Westenfeld R. The K‑factor in chronic kidney disease: biomarkers of calcification inhibition and beyond. Nephrol Dial Transplant 2014, 29:1267–70. [ Links ]
10. Price PA, Faus SA, Williamson MK. Warfarin‑induced artery calcification is accelerated by growth and vitamin D. Arterioscler Thromb Vasc Biol. 2000, 20:317–27. [ Links ]
11. Malabu UH, Manickam V, Kan G, Doherty SL, Sangla KS. Calcific uremic arteriolopathy on multimodal combination therapy: Still unmet goal. Int J Nephrol 2012; 390768, 2012 [ Links ]
12. Nigwekar SU, Brunelli SM, Meade D, Wang W, Hymes J, Lacson E. Sodium Thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013,8:1162–70. [ Links ]
Filipa Brito Mendes
Nephrology Department
Algarve Hospital Centre, Faro, Portugal
E‑mail:filipabritomendes@gmail.com
Authors Contributions:
Filipa Brito Mendes: manuscript writing
Sofia Couto Rocha: grammatical correction and abstract writing
Rodica Agapii, André Fragoso, Teresa Jerónimo, Ana Pimentel: clinical and laboratory research
Ana Silva and Pedro Leão: project orientation and critical analysis
Disclosure of potential conflicts of interest: None declared
Received for publication: Dec 13, 2015
Accepted in revised form: May 16, 2016


{kind=link}