










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.2 Lisboa jun. 2017
CASE REPORT
Recurrence of anti‑factor H associated C3 glomerulonephritis in the kidney allograft
Rita Magriço1, Fernanda Carvalho2, António Pedro Alves de Matos3, Cristina Jorge4, Ana Mateus1, Pedro Cruz1, Carlos Oliveira1, Manuel Amoedo5, Fernando Nolasco2, Aura Ramos1
1 Serviço de Nefrologia do Hospital Garcia de Orta, Almada, Portugal
2 Laboratório de Morfologia Renal‑Servico de Nefrologia, Hospital Curry Cabral – CHLC, Lisboa, Portugal
3 Centro de microscopia electrónica e histopatologia Egas Moniz, Campus Universitário, Caparica, Portugal
4 Serviço de Nefrologia, Hospital de Santa Cruz – CHLO, Carnaxide, Portugal
5 Serviço de Nefrologia, Hospital do Espírito Santo de Évora, Évora, Portugal
ABSTRACT
C3 glomerulonephritis is a rare disease defined by deposits of C3 on immunofluorescence due to dysregulation of the alternative pathway of the complement cascade. We present a case of a female patient that presented with nephrotic syndrome at the age of 18 due to C3 glomerulonephritis and progressed to end‑stage renal disease after 9 years. She lost the first kidney graft on the third day after transplantation due to vascular thrombosis. A second kidney transplant was performed when the patient was 30 years old.
C3 glomerulonephritis recurred on the kidney graft and the patient returned to dialysis 10 years after the transplant. Study of the alternative complement pathway revealed anti‑factor H antibodies. She is currently on evaluation for a third kidney transplant and there is no consensus on the best treatment strategy. We present the literature available for treatment of C3 glomerulonephritis and the therapeutic options for this patient.
Key Words: Alternative complement pathway, complement C3, complement factor H, glomerulonephritis, kidney diseases, transplants.
INTRODUCTION
C3 glomerulonephritis is a rare disease defined by deposits of C3 on immunofluorescence due to dysregulation of the alternative pathway of the complement cascade. Recurrence rate after transplantation is high and is associated with poor renal survival. Therapeutic strategies capable of changing the natural course of the disease are ill defined.
CASE REPORT
We report a case of a 18‑year‑old Caucasian female with a past history of allergic rhinitis that was evaluated for the first time in a nephrology consultation in 1993 due to nephrotic syndrome. The patient reported no significant symptoms and clinical examination was unremarkable. There was no family history of renal disease. Albuminaemia was 1.86 g/dL and 24‑hour proteinuria was 4.1g. Haemoglobulin, platelet and white cell count were normal. Serum creatinine was 0.6 mg/dL. Total complement activity (CH50) was decreased and complement C3 and C4 were also low.
Anti‑nuclear antibodies (ANAs) were positive but anti‑double stranded DNA antibodies (anti‑dsDNA) were negative. Renal biopsy disclosed a membranoproliferative pattern (Figure 1) with C3 deposits (Figure 2) and minimal immunoglobulin deposits. Electron microscopy (performed a posteriori) revealed discontinuous deposits (Figure 3) located on the capillary wall (subendothelial, intramembranous and subepithelial) as well as on the mesangium with low density.
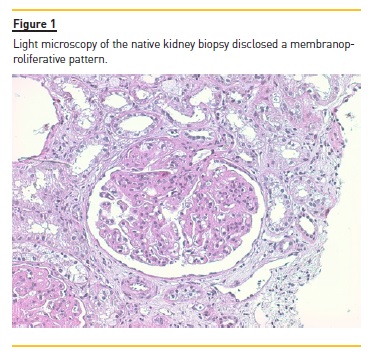
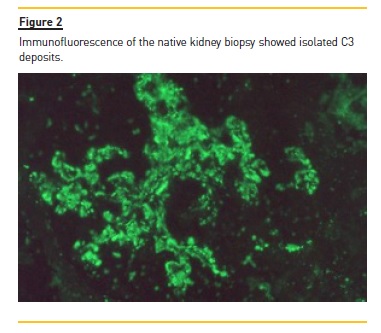
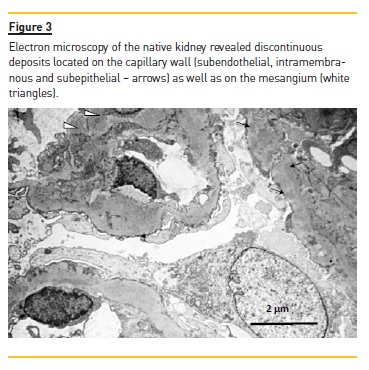
Kidney disease progressed until the patient started peritoneal dialysis in 2002. In 2003 the patient received the first kidney graft but transplantectomy was performed on the third day after transplantation due to vascular thrombosis of the graft.
In 2005 the patient developed hypertension. In February 2005 she presented with vomiting and seizures and was admitted to the hospital. Venous cerebral thrombosis was detected. Extensive testing for thrombophilia was negative (including VDRL, TPHA, deficiency of antithrombin III, protein C and protein S, lupus anticoagulant, antiphospholipid and anticardiolipin antibodies; homocysteine levels were normal). At this time ANAs were negative and C3 and C4 were normal. Search for mutations of factor II (prothrombin) and factor V Leiden mutation were performed a posterior and were negative. At discharge, the patient did not present any neurological deficit. She started oral anticoagulation with warfarin. A statin was also introduced.
In October 2005 the patient received a second kidney transplant. The donor was a 28‑year‑old male whose death resulted from polytraumatism. The serum creatinine of the donor was 0.6mg/dL. The donor had 3 HLA compatibilities with the recipient (1 on HLA‑B and 2 on HLA‑DR).
The panel reactive antibody of the recipient was 0%. Cold ischaemia time was 15h. Immunosuppression was performed with thymoglobulin (1.5mg/Kg for 7 days), prednisone (maintenance dose of 5mg/day), mycophenolate (maintenance dose of 500 mg twice a day) and tacrolimus (trough target levels of 8‑12ng/mL in the first year, 6‑8ng/mL between the first and second years and 4‑6ng/mL thereafter).
The patient presented with microscopic haematuria since the first month after the transplant and proteinuria since the end of the first year after the transplant.
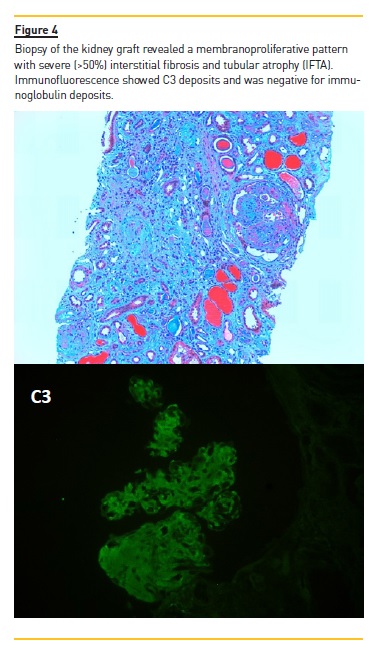
She was treated with anti‑hypertensive drugs including angiotensin‑converting enzyme (ACE) inhibitors, despite which proteinuria persisted. Between 2007 and 2011 she was admitted to hospital 4 times due to gluteal abscess, treated with antibiotics and surgical drainage. No other complications occurred. Kidney function remained normal (serum creatinine 0.8mg/dL) until the sixth year post transplant (2011), after which there was a slow but progressive decrease in renal function. In 2014, proteinuria became nephrotic (urine protein to creatinine ratio increased from 2.3 g/g in 2013 to 4 g/g in 2014) and renal function deteriorated (increase in serum creatinine from 1.9 mg/dL to 3.2 mg/dL). Monoclonal gammopathy was excluded and testing for ANAs, anti‑dsDNA and anti‑neutrophil cytoplasmic antibodies (ANCAs) were negative. Biopsy of the kidney graft (Figure 4) revealed a membranoproliferative pattern with C3 deposits on immunofluorescence (no immunoglobulin deposits were detected).
There was severe (>50%) interstitial fibrosis and tubular atrophy (IFTA). Recurrence of C3 glomerulonephritis was diagnosed but due to extensive renal damage on biopsy and lack of robust evidence on effective treatment, immunosuppression was not increased. In 2015 the patient was admitted to hospital with skin lesions. Diagnostic work‑up revealed positive cryoglobulins.
Skin biopsy disclosed calciphylatic skin lesions. Due to uraemic syndrome, the patient restarted peritoneal dialysis in 2015.
In 2016 the patient requested evaluation for a third kidney transplant. She presented with low C3, normal CH50 and normal AH50 (alternative pathway haemolytic activity). Genetic study of the complement was normal.
Serum levels of C1q, C2, C6, C7, C9, Factor H and Factor I were slightly elevated (up to 1.36 times the maximum value of the reference range). Anti‑H antibodies were detected (205 UA/mL, reference value < 27). She is currently 41 years old. Because there is no consensus on the best treatment strategy to adopt in this case, we present the literature available for treatment of C3GN.
DISCUSSION
We present a case of a patient with C3GN. She presented with nephrotic syndrome at the age of 18 and progressed to end‑stage renal disease after 9 years. She lost the first kidney graft on the third day after transplantation due to vascular thrombosis. A second kidney transplant was performed when the patient was 30 years old. C3GN recurred on the kidney graft and the patient returned to dialysis 10 years after the transplant. Study of the alternative complement pathway revealed anti‑factor H antibodies. She is currently on evaluation for a third kidney transplant and there is no consensus on the best treatment strategy.
The term C3 glomerulopathies includes two diseases (dense deposit disease – DDD – and C3 glomerulonephritis – C3GN), both characterized by C3 glomerular deposits on immunofluorescence with minimal or absence of immunoglobulin deposits1. C3 glomerulopathies are considered an independent entity because the definition signals a disturbance of the alternative pathway of the complement, with important implications for the diagnostic work‑up, therapy and prognosis.
Whilst in DDD the deposits are intramembranous, with high density and involve large segments of the glomerular basement membrane, in C3GN deposits are less osmiophilic and are mostly mesangial and subendothelial, although there may also be intramembranous and subepithelial deposits2.
C3 glomerulopathies are characterized by excessive activation of the C3 convertase of the alternative pathway of the complement cascade. This patient had low C3 and low CH50 due to consumption of the complement components and increased C5b‑9 because of increased complement activity. Complement test results may be normal when testing is not performed in the acute phase of the disease. The activity of the C3 convertase may be increased due to genetic defects or auto‑antibodies against the regulators of C3 convertase (the main regulators are factor H, I and B).
Auto‑antibodies may also increase the activity of the complement by binding and stabilizing C3 convertase (C3 nephritic factor – C3Nef). Diagnostic investigation of C3 glomerulopathies therefore requires genetic study of mutations of the complement and detection of auto‑antibodies.
This distinction is important, since genetic defects are treated with periodic infusions of plasma whereas depletion of auto‑antibodies is performed with plasma exchange and immunosuppression. It should be noted that genetic defects may coexist with auto‑antibodies and that several antibodies may be present simultaneously in a single patient. In a study of 17 patients with alternative pathway‑mediated Glomerulopathies positive for anti‑factor H antibodies, 7 also had C3Nef3. Finally, it is also important to exclude auto‑antibody formation due to monoclonal gammopathy, in which case therapy is directed at the haematological disease. Systematic description of diagnostic work‑up of disorders of the alternative pathway of the complement is available4, as is contact information on Portuguese laboratories where complement investigation can be performed5.
C3 glomerulonephritis was reported for the first time in 20076 and there are very few cases reported of this recently defined and rare disease. Recurrence rate after transplantation is greater than 50%7 and first urinary abnormalities are detected one to two years after transplantation7.
Renal survival after 10 years is approximately 50%8.
From the cases reported in the literature of C3 glomerulopathy associated with factor‑H antibodies, the authors selected three that correspond to patients with C3GN where immunosuppression was attempted to change the natural course of the disease. One female patient presented with nephritic syndrome at the age of 16, and progressed to end‑stage renal disease after 3 years despite treatment with corticosteroids. Recurrence of disease caused return to dialysis 7 months after the first kidney transplant and after 1 year of the second transplant despite treatment with plasma exchange and rituximab9. Another patient was a 36‑year‑old pregnant woman with sinusitis who presented with acute renal failure and nephrotic syndrome.
Despite corticosteroids and cyclophosphamide, she started dialysis that same year. Disease recurred on the first kidney graft one month after transplantation and the patient performed plasma exchange, rituximab and switch from cyclosporine to tacrolimus with transient improvement until worsening of proteinuria and renal function on the first year after transplant.
At this time, immunosuppression was not increased and the patient returned to dialysis three years after transplantation. A second transplant was performed and despite plasma exchange, corticosteroids and cyclophosphamide, the kidney graft was lost 8 months after transplantation. Biopsy of the second graft disclosed thrombotic microangiopathy associated with the same anti‑factor H antibody10. The third patient was a 5‑year‑old boy with proteinuria and normal renal function that was found to have three combined abnormalities in the regulation of the complement: anti‑factor H antibodies, C3Nef and a mutation on factor I. Despite treatment with corticosteroids, rituximab and mycophenolate, proteinuria progressed (from 1.0 to 5.3g/day) and eculizumab was started with proteinuria decrease. Transient interruption of the drug resulted in worsening of proteinuria (1.7 to 9.3 g/day) which ameliorated when eculizumab was resumed. On the third year of follow‑up the patient remained dependent on every other week administration of 1200 mg of eculizumab. He maintained normal renal function and proteinuria <2g/day11.
The variability of clinical manifestations of patients with alternative pathway related diseases depends on the specific abnormality present on complement regulation. Anti‑factor H antibodies cause a significant proportion of cases of atypical haemolytic uraemic syndrome (aHUS) and a small proportion of cases of C3GN12. Differences between patients on the binding site on factor H and on the affinity of the antibody to factor H have been appointed to explain the clinical variability observed3. The simultaneous presence of various defects on complement regulation is considered to be an additional contributing factor3.
In‑depth knowledge of the cause of complement dysregulation in each individual patient is important to choose the most appropriate treatment strategy and the parameters that can be monitored to assess treatment response.
The conclusions of a KDIGO controversies conference on treatment of C3 glomerulopathies were published in 201713. If a third transplant is considered in this patient, it must not be performed during acute inflammation and risk of recurrence has to be considered13.
Treatment recommendations include target blood pressure ≤120/80 mmHg, adequate nutrition and lipid control for all patients. In moderate disease (defined by proteinuria ≥ 0.5g/day, increase in creatinine or moderate inflammation on biopsy), prednisone and mycophenolate are recommended13. Given that these therapeutic measures were followed and the outcome of this patient (graft survival was similar to native kidney survival, suggesting little change on the natural course of disease with therapy), these recommendations are insufficient. In severe disease (defined by urinary protein ≥ 2g/day or maintenance of inflammation or creatinine increase despite the abovementioned therapy), there is no known strategy to reduce risk of recurrence (13, supplementary table 10) and data is insufficient to recommended use of eculizumab13.
In conclusion, C3GN is associated with decreased renal survival. Although knowledge of the pathophysiology of the disease has increased significantly, treatment strategies capable of changing the natural course of the disease are lacking.
References
1. Pickering M, DAgati V, Nester C. C3 glomerulopathy: consensus report. Kidney Int. 2013 Dec; 84(6):1079‑89. [ Links ]
2. Sethi S, Haas M, Markowitz GS, et al. Mayo Clinic/Renal Pathology Society consensus report on pathologic cassification, diagnosis, and reporting of GN. J AmSoc Nephrol. 2016; 27:1278–87. [ Links ]
3. Blanc C, Togarsimalemath SK, Chauvet S, et al. Anti–Factor H autoantibodies in C3 glomerulopathies and in atypical hemolytic uremic syndrome: one target, two diseases. J. Immunol. 2015; 194:5129–38. [ Links ]
4. Angioi A, Fervenza FC, Sethi S, et al. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016 Feb; 89(2):278‑88. [ Links ]
5. Carvalho F, Nolasco F. C3 glomerulopathies: a new category encompassing rare complement mediated glomerulonephritis. Port J Nephrol Hypert. 2016; 30(4):239‑45. [ Links ]
6. Servais A, Fremeaux‑Bacchi V, Lequintrec M. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007 Mar; 44(3):193‑9. [ Links ]
7. Zand L, Lorenz E, Cosio F, et al. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol. 2014 May; 25(5):1110‑7. [ Links ]
8. Servais A, Noel L, Roumenina L, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012 Aug; 82(4):454‑64. [ Links ]
9. Goodship THJ, Pappworth IY, Tothb T, et al. Factor H autoantibodies in membranoproliferative glomerulonephritis. Mol Immunol. 2012 Oct; 52(3‑4):200‑6. [ Links ]
10. Lorcy N, Rioux‑Leclercq N, Lombard ML, Le Pogamp P, Vigneau. Three kidneys, two diseases, one antibody? Nephrol Dial Transplant. 2011; 26:3811–33.
11. Payette A, Patey N, Dragon‑Durey MA, Fremeaux‑Bacchi V, Le Deist F, Lapeyraque AL. A case of C3 glomerulonephritis successfully treated with eculizumab. Pediatr Nephrol. 2015; 30:1033–7. [ Links ]
12. Dragon Durey MA, Sinha A, Togarsimalemath SK, Bagga A. Anti‑complement>‑factor H‑associated glomerulopathies. Nat Rev Nephrol. 2016 Sep; 12(9):563‑78. [ Links ]
13. Goodship THJ, Cook HT, Fakhouri F. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017; 91(3):539‑51. [ Links ]
Rita Magriço
Rua Luís Pastor de Macedo, Lote 17/18 3ºD
1750‑159 Lisboa
E-mail: rita.magrico@yahoo.com
ACKNOWLEDGEMENTS
The authors acknowledge Dr Karina Soto, the Nephrology Collaborative Group of the Mayo Clinic, the Nephrology Department of Hospital Fernando Fonseca and the Lisbon Nephropathology Group (GNL) for the opportunity to present and discuss this case report on the Lisbon Clinical Nephrology Update.
Contributions of the authors to the article:
RM assembled the data and wrote the manuscript. FC performed the histological diagnosis. AP Alves Matos took the photographs of the electron microscopy analysis. CJ evaluated the patient for third transplant and requested the genetic and immunological study of the alternative pathway of the complement. AM, PC and CO followed the patient during the second renal transplant. MA followed the patient until she received the second kidney transplant. The manuscript was reviewed by all the authors.
Disclosure of potential conflicts of interest: none declared
Received for publication: Jun 10, 2017
Accepted in revised form: Jun 19, 2017


{kind=link}