










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.2 Lisboa jun. 2017
CASE REPORT
Severe Systemic Lupus Erythematosus presentation in patient with alternative complement pathway mutations
Fernando Pereira1, Liliana Cunha1, Pedro Campos1, Ana Gaspar1, Rita Manso2, Karina Soto1
1 Serviço de Nefrologia, Hospital Prof. Doutor Fernando Fonseca EPE, Amadora, Portugal
2 Serviço de Anatomia Patológica, Hospital Prof. Doutor Fernando Fonseca EPE, Amadora, Portugal
ABSTRACT
Systemic lupus erythematosus (SLE) is an autoimmune disease which can involve almost any organ, making its difficult therapeutic approach. Immune complex deposition can often activate complement, accounting for many of SLE clinical manifestations and laboratory findings.
We present a case of a patient who presented with acute pancreatitis and acute kidney injury as onset manifestations of SLE, later developing neurological manifestations, who was successfully treated with rituximab, plasma exchange and steroids as induction therapy. Persistently low C3 level led to a genetic analysis of the complement system components. We found three polymorphisms in the alternative pathway of complement regulators (complement factor H c2669 G>T, p.Ser890Ile and c3019 G>T, p.Val1007Leu and complement factor I c.482+6 G>T), two of which have been correlated with atypical haemolytic uraemic syndrome and dense deposit disease and also complement factor H‑related protein (CFHR1 and CFHR3) mutations by deletion. This raises the question whether these polymorphisms and mutations played any role in our patients clinical course.
Key‑Words: SLE, complement, complement factor H, rituximab, plasma exchange, lupus pancreatitis
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disease with broad clinical manifestations characterized by the production of autoantibodies1 which can have multi‑organ involvement2 with tissue damage produced mainly by complement activation by immune complexes (IC).3 As a result of this activation, patients present hypocomplementaemia during disease flares, but C3 and C4 levels are expected to return to normal range between flares. Complement alternative pathway activation with C3 deficiency has been reported in SLE manifestations, notably lupus nephritis.4,5 However, this association is unclear, as partial deficiencies of complement proteins have not been associated with SLE.6,7
Inherited deficiencies of several complement proteins strongly predispose to SLE while mutations of complement inhibitors are found mostly in kidney diseases such as atypical haemolytic uraemic syndrome (aHUS).
Herein we present a very challenging case of severe SLE with multisystem involvement with C3 levels persistently below normal range and mutations of complement regulators of the alternative pathway, raising further considerations regarding treatment approach.
CASE REPORT
An African‑native 45‑year‑old woman was admitted to the emergency department with dyspnoea, fever (38.0°C), headache, abdominal pain and diarrhoea over the course of the previous two days. She also complained of sore throat, foamy urine and low urine output over the last few days. She reported complaints of low‑grade fever (37.5 ± 37.8°C), malaise, anorexia during the previous month and weight loss of approximately 35kg in the last two months. She had been previously medicated with non‑steroidal anti‑inflammatory drugs and amoxicillin‑clavulanic acid. On observation, she was sleepy (GCS 12), hypotensive (BP 86/42mmHg, HR 102ppm), with red throat and diffuse abdominal pain. Laboratory study revealed haemoglobin 7.9g/dL, WBC 5.6x10/L, platelet count 109x10/L, AST 89UI/L, ALT 34UI/L, ALP 261UI/L, GGT 405UI/L, LDH 517UI/L, total bilirubin 0.29mg/dL), serum amylase 3096 UI/L, serum albumin 1.13g/dL, serum creatinine (sCr) 6.17mg/dL, urea 116mg/dL, triglycerides 515mg/dL, total cholesterol 179mg/dL and raised inflammatory markers: CRP 6.96mg/dL, ESR 140mm. Her urine showed haematuria and proteinuria (Hb 2+, Prot >400mg/dL, albumin‑creatinine ratio in urine (ACR) 7300mg/g). An abdominal CT was performed and confirmed acute pancreatitis (Figure 1).
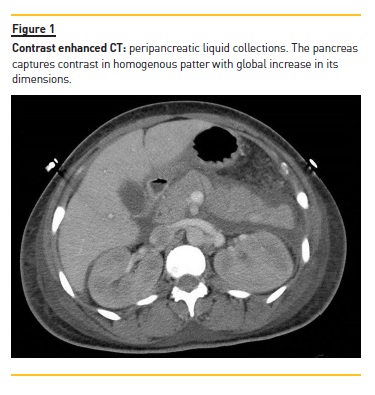
Immunology studies showed antinuclear antibodies 1/320, raised anti‑dsDNA antibodies 78 UI/mL, positive anti‑RPN, and extremely low C3 and C4 complement fraction (41, 6.4 mg/dL respectively). We ruled out thrombotic microangiopathy since she had normal haptoglobin level (131mg/dL) and the peripheral blood smear did not show any schizocytes. Also, serology for HIV, Hepatitis B and C, Epstein‑Barr Virus, CMV and syphilis, blood and urine cultures and search for Mycobacterium tuberculosis and Plasmodium falciparum were all negative.
The diagnosis of SLE was therefore established with severe renal, haematological and pancreatic involvement.
The patient rapidly developed thrombocytopaenic anaemia, oligoanuria and tetraparesis with nystagmus and opsoclonus. Brain MRI showed hyperintense signal(long TR and T2) on the right side of the brainstem and medium cerebellar stalk. EMG showed predominantly motor polyneuropathy. Renal replacement therapy was started together with intravenous methylprednisolone 500mg pulses (3 days) followed by oral prednisolone (PDN 60mg od). As haematological and neurological disorders progressed, with very low platelet count (21000/uL), weekly rituximab (RTX) was started (375mg/m2 of body surface area) on a total of 4 doses.
One day later (at 9th day of admission) plasma exchange (PE) was started, undergoing 5 PE treatments. Kidney function rapidly improved, stopping haemodialysis at the 5th day after RTX treatment. Haematological, neurological and pancreatic improvement was evident after PE (3rd week of entire treatment).
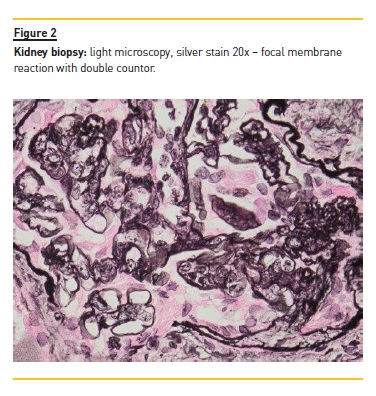
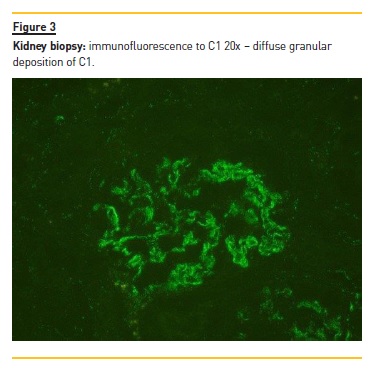
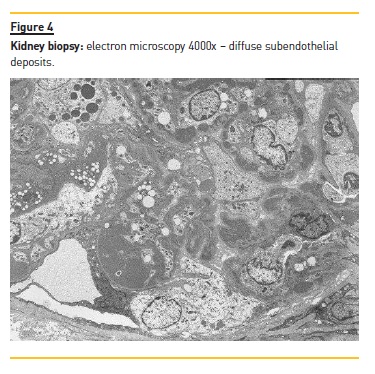
Kidney biopsy was performed on the 30th day of admission and showed an active, diffuse, global, proliferative glomerulonephritis classified as class IV‑G Lupus Nephritis (ISN/RPS 2004 classification) with activity index of 4/24 and chronicity index of 1/12 (Figure 2). Immunofluorescence (IF) showed granular mesangial and pseudolinear membranous deposits of IgG (+++), C3 (+++), C1 (+++), IgA (++) and IgM (+) (Figure 3) and electron microscopy (EM) showed electron dense subendothelial immune deposits, resembling dense deposit disease (DDD) (Figure 4).
Mycophenolate mofetil (MMF) was started on the 31st day of admission (1000mg bid), tapering PDN to 30mg od at discharge, two months after admission.
Laboratory findings at discharge were Hb 9.5g/dL, platelet count 371x10/L, sCr 1.10mg/dL, ACR 910mg/g, normal C3 and C4 (93, 14.3mg/dL) and negative dsDNA (9.7UI/mL).
During follow‑up, C3 levels have remained below the normal range, despite there being no signs of SLE flare. Therefore, complement molecular study was performed in an attempt to identify abnormalities in the alternative complement pathway. Genetic analysis revealed that the patient carried two mutations in heterozygosis of complement factor H (CFH) gene (c2669 G>T, p.Ser890Ile and c3019 G>T, p.Val1007Leu) and 1 mutation in heterozygosis of Factor I (CFI) gene (c.482+6 G>T). Also, mutations by deletions on complement factor H‑related proteins (CFHR), CFHR1 and CFHR3 in heterozygosis.
At 30 month follow‑up patient complains only of peripheral polyneuropathy, remaining in total remission with creatinine clearance of 129.9mL/min, proteinuria of 0.814g/day, and ACR 206mg/g, with normal urine sediment, with no haematological abnormalities or autoantibodies. Treatment remained based on MMF and hydroxychloroquine until two years of follow‑up.
DISCUSSION
The patient presented here had a severe multiorgan presentation of SLE with gastrointestinal, renal, haematological and neurological involvement. Acute pancreatitis is a rare manifestation of SLE.8‑13
Pathophysiology is unclear but available evidence suggests that vasculitis, microthrombus formation, anti‑pancreatic antibodies and inflammation due to T‑cell
Infiltration and complement activation might be involved.13 Neuropsychiatric systemic lupus erythematosus (NPSLE) is the least understood but probably the most prevalent manifestation of lupus. Its pathogenesis is multifactorial and involves various inflammatory cytokines, autoantibodies, and ICs resulting in vasculopathic, cytotoxic and autoantibody‑mediated neuronal injury and is associated with increased morbidity and mortality.14
Our patient presented with light neurological changes but developing rapidly severe brainstem involvement, nystagmus and cognitive impairment. Haematological abnormalities are common findings in patients with SLE. Low platelet count is mainly associated to neuropsychiatric manifestation, haemolytic anaemia, and kidney disease.15 Thrombocytopenia can be regarded as an important prognostic indicator of survival in patients with SLE. Our patient presented with severe thrombocytopaenia and severe anaemia, but no signal of haemolysis was found. Additionally, the patient developed severe acute kidney injury due to class IV‑G lupus nephritis with full house immune deposits, but with C3 (+++), no C4 on IF and EM showing electron‑dense deposits resembling DDD pattern. As serum C3 level persisted low, the patient was genetically studied demonstrating 2 CFH and 1 CFI mutations, associated with deletion on CFHR1 and CFHR3. Complement factors consumption during SLE flares exacerbations is a hallmark of this syndrome. In fact, low C3 and C4 and a high dsDNA titre can preclude renal flare.16 CFH is the main regulator of the alternative pathway of the complement system and regulates complement activation both in fluid phase and on cellular surfaces.17
Mutations and polymorphisms in CFH gene have been found to be associated with aHUS, DDD and age‑related macular degeneration. The two mutations identified in our patient in CFH have been repeatedly reported to be associated with aHUS, but have also been found in the other two conditions.18,19
Many of these CFH genetic variations disrupt the regulatory role of factor H. However, data illustrate that these amino acid substitutions in those mutations do not influence the expression/secretion of FH.20 Notwithstanding, the same authors concluded that associations of complement regulation abnormalities could explain the persistent complement alternative pathway activation.
Our patient has a CFI mutation with unknown effect to our knowledge, and moreover she also carries CFHR1 and CFHR3 deletion. It was described that these mutations encode a CFH protein with reduced capacity to regulate the activity on cellular surfaces being associated with an increased risk of aHUS.19 The latter observation emphasizes that multiple concurrent factors may be necessary in individual patients for alternative pathway disease manifestation. A recently published study showed that CFH deficiency accelerates development of lupus nephritis in an accurate mouse model of human SLE (MRL‑lpr mice), sharing many features of SLE in terms of production of autoantibodies and consumptive hypocomplementemia.21 Pathologic findings in these animals included marked glomerular subendothelial, mesangial and subepithelial deposition of IC containing C3.
It is remarkable that a SLE patient with severe systemic involvement, particularly severe lupus nephritis with C3 deposition similar to that of DDD, also carried all these genetic mutations, raising the question whether they may play any role in our patients clinical presentation.
However, it is also known that there is a high allelic frequency of these two CFH variants in sub‑Saharan African populations20, supporting the hypothesis of this being an incidental finding.
Our patient showed a good response to PE associated with RTX and steroids as induction treatment.
Although randomized controlled trials (RCTs) have failed to show superiority of rituximab over standard immunosuppression alone, in reducing clinical activity and achieving complete or partial remission without subsequent flares22,23, data from one of these trials (LUNAR) showed a statistical significant difference in the number of patients reaching proteinuria remission at 78‑weeks.24,25 Moreover, these two trials excluded patients with severe or refractory SLE, such as the case presented, with severe gastrointestinal, neurological, haematological and renal involvement, which might be a reason to consider this therapeutic option. Regarding patients with SLE with NPSLE, a recent review of published data of patients treated with RTX found an 85% response rate with partial or complete remission.26 Regarding PE there is lack of evidence in lupus nephritis treatment. However, the American Society for Apheresis accepts it as second‑line therapy to treat SLE manifestations, such as NPSLE.27 It has been proposed that PE modulates humoral components of immune response, removing autoantibodies and IC. Despite no RCTs showing its efficacy in SLE patients, there has been case series published showing evidence of its utility, mainly in NPSLE, simultaneously with other immunosuppressive treatment.28
In summary, we report a severe SLE case with acute pancreatitis, NPSLE, severe bicytopenia and lupus nephritis, who carries different complement alternative pathway mutations. To our knowledge, this is the first case published with this association. The specific role of these findings is not yet clarified. Nevertheless, we achieved a complete prolonged remission with treatment with RTX, PE and steroids as induction treatment and MMF as maintenance therapy. Early diagnosis and treatment probably played a major role in the favourable outcome of this serious and rapidly deteriorating SLE case.
References
1. Mills JA. Systemic lupus erythematosus. N Engl J Med. 1994; 330: 1871–1879 [ Links ]
2. Wallace DJ. The clinical presentation of systemic lupus erythematosus. In: Wallace DJ, Hahn BH, editors. Dubois lupus erythematosus. 5th ed. Baltimore: Lippincott Wilkins & Wilkins. 1997; 627 [ Links ]
3. Birmingham DJ, Irshaid F, Nagaraja HN et al. The complex nature of serum C3 and C4 as biomarkers of lupus renal flare. Lupus. 2010; 19: 1272–1280 [ Links ]
4. Tsukamoto H, Horiuchi T, Kokuba H et al. Molecular analysis of a novel hereditary C3 deficiency with systemic lupus erythematosus. Biochem Biophys Res Commun. 2005; 330: 298–304 [ Links ]
5. Nozal P, Garrido S, Martinez‑Ara J et al. Case report: Lupus nephritis with autoantibodies to complement alternative pathway proteins and C3 gene mutation. BMC Nephrology. 2015; 16: 40 [ Links ]
6. Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007; 40: 560–566 [ Links ]
7. Kościelska‑Kasprzak K, Bartoszek D, Myszka M et al. The complement cascade and renal disease. Arch Immunol Ther Exp. 2014; 62: 47–57 [ Links ]
8. Reifenstein E, Reifenstein EJ, Reifenstein G. A variable symptom complex of undetermined etiology with fatal termination. Arch Intern Med 1939; 65: 553–574 [ Links ]
9. Reynolds JC, Inman RD, Kimberly RP et al. Acute pancreatitis in systemic lupus erythematosus: report of twenty cases and a review of the literature. Medicine. 1982; 61: 25–32 [ Links ]
10. Hiraishi H, Konishi T, Ota S et al. Massive gastrointestinal hemorrhage in systemic lupus erythematosus: successful treatment with corticosteroid pulse therapy. Am J Gastroenterol 1999; 94: 3349–3353 [ Links ]
11. Carducci M, Calcaterra R, Mussi A et al. Acute pancreatitis as initial manifestation of systemic lupus erythematosus and subacute cutaneous lupus erythematosus: report of two cases. Lupus. 2008; 17: 695–697 [ Links ]
12. Jia Y, Ortiz A, Mccallum R et al. Acute pancreatitis as the initial presentation of systematic lupus Erythematosus. Case Reports in Gastrointestinal Medicine; 2014. Article ID 571493 [ Links ]
13. Wang F, Wang NS, Zhao BH et al. Acute pancreatitis as an initial symptom of systemic lupus erythematosus: a case report and review of the literature. World J Gastroenterol. 2005; 11: 4766–4768 [ Links ]
14. Popescu A, Kao AH. Neuropsychiatric systemic lupus erythematosus. Current Neuropharmacology. 2011; 9: 449–457 [ Links ]
15. Fayyaz A, Igoe A, Kurien BT, et al. Haematological manifestations of lupus. Lupus Science & Medicine. 2015; 2: e000078. [ Links ]
16. Birmingham DJ, Irshaid F, Nagaraja HN et al. The complex nature of serum C3 and C4 as biomarkers of lupus renal flare. Lupus. 2010; 19: 1272–1280 [ Links ]
17. Pangburn MK, Schreiber RD, Muller‑Eberhard HJ. C3b deposition during activation of the alternative complement pathway and the effect of deposition on the activating surface. J Immunol. 1983; 131: 1930–1935 [ Links ]
18. Moore I, Strain L, Pappworth I et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010; 115: 379 –87 [ Links ]
19. Maga TK, Nishimura CJ, Weaver AE et al. Mutations in alternative pathway complement proteins in American patients with atypical haemolytic uremic syndrome. Hum Mutat. 2010; 31: E1445–1460 [ Links ]
20. Tortajada A, Pinto S, Martinez‑Ara et al. Complement factor H variants I890 and L1007 while commonly associated with atypical haemolytic uremic syndrome are polymorphisms with no functional significance. Kidney Int. 2012; 81: 56–63
21. Bao L, Haas M, Quigg RJ. Complement factor H deficiency accelerates development of lupus nephritis. J Am Soc Nephrol. 2011; 22: 285–295 [ Links ]
22. Merrill JT, Neuwelt CM, Wallace DJ et al. Efficacy and safety of rituximab in moderately to severely active systemic lupus erythematosus: the randomized, double‑blind, phase
II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010; 62: 222–233
23. Rovin BH, Furie R, Latinis K et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab Study. Arthritis Rheum. 2012; 64: 1215–26 [ Links ]
24. Lightstone L. The landscape after LUNAR: rituximabs crater‑filled path. Arthritis Rheum. 2012; 64: 962–965 [ Links ]
25. Gunnarsson I, Jonsdottir T: Rituximab treatment in lupus nephritis – where do we stand? Lupus. 2013; 22:381–389 [ Links ]
26. Narvaez J, Rios‑Rodriguez V, de la Fuente D et al. Rituximab therapy in refractory neuropsychiatric lupus: current clinical evidence. Semin Arthritis Rheum. 2011; 41: 364–372 [ Links ]
27. Schwartz J, Winters JL, Padmanabhan A et al. Guidelines on the use of therapeutic apheresis in clinical practice‑evidence‑based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013; 28: 145–284 [ Links ]
28. Bartolucci P, Brechignac S, Cohen P et al. Adjunctive plasma exchanges to treat neuropsychiatric lupus: a retrospective study on 10 patients. Lupus. 2007; 16: 817–822 [ Links ]
Correspondence to:
Fernando Pereira
Nephrology Department
Hospital Prof. Doutor Fernando Fonseca EPE
IC 19, 2720‑276 Amadora, Portugal
E-mail: fernando.g.pereira@hff.min‑saude.pt
Disclosure of potential conflicts of interest: none declared
Received for publication: Jun 11, 2017
Accepted in revised form: Jun 19, 2017

