










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.3 Lisboa set. 2018
CONSENSUS DOCUMENT
Portuguese consensus document statement in diagnostic and management of atypical hemolytic uremic syndrome
Ana Azevedo1, Bernardo Faria2, Catarina Teixeira3, Fernanda Carvalho4, Gisela Neto5, Josefina Santos6,7, Maria do Céu Santos8, Nuno Oliveira9,10, Teresa Fidalgo11, Joaquim Calado12,13
1 Nephrology Consultant in Hospital Vila Franca de Xira
2 Nephrology Department, Hospital de Braga
3 Nephrology Department, Hospital Beatriz Ângelo, Loures
4 Nephropathologist in Nefrolab-Clara Saúde, Paço de Arcos
5 Pediatric Nephrology Unit, Department of Women, Children and Adolescents – Hospital de Dona Estefânia – Centro Hospitalar de Lisboa Central
6 Nephrology Department, Hospital de Santo António – Centro Hospitalar do Porto
7 Unit for Multidisciplinary Research in Biomedicine, Instituto de Ciências Biomédicas Abel Salazar (ICBAS) – Universidade do Porto
8 Immunobiologist in Clinical Pathology Department; Hospital de São José – Centro Hospitalar de Lisboa Central
9 Nephrology Department, Centro Hospitalar Universitário de Coimbra
10 University Clinic of Nephrology, Faculty of Medicine – Universidade de Coimbra
11 Molecular Haematology Laboratory, Department of Clinical Hematology, Centro Hospitalar Universitário de Coimbra
12 Nephrology Department, Hospital Curry Cabral – Centro Hospitalar de Lisboa Central
13 Centre for Toxicogenomics and Human Health (ToxOmics), Genetics, Oncology and Human Toxicology, Nova Medical School – Universidade Nova de Lisboa
ABSTRACT
Among thrombotic microangiopathies (TMA), the hemolytic uremic syndrome associated with dysregulation of the alternative complement pathway (aHUS) is one of the most challenging diseases a nephrologist can face. By the end of the XXth century, the complements role was unraveled with the discovery that mutations in the factor H coding gene were responsible for aHUS. But it was the acknowledgment that pharmacological C5-9 blockage provided a cure for aHUS that fostered the interest of the nephrology community in the genetics, pathophysiology and therapeutics of, not only of aHUS, but TMA in general. The molecular genetics of aHUS is technically demanding and, as such, in Portugal (alike many other European countries) a single laboratory emerged as a national reference center. The fact that all samples are evaluated in a single center provides a unique opportunity for data collection and a forum for discussion for all those interested in the field: immunologists, molecular geneticists, pathologists and nephrologists. The current consensus document emerged from such a discussion forum and was sponsored by the Portuguese Society of Nephrology. The goal is more to portray the Portuguese picture regarding the diagnostic approach and therapeutic options than to extensively review the state of the art of the subject. The accompanying documents that are published as supplementary data are in line with that goal. They range from the informed consent and clinical form to be sent together with the biological samples for genetic testing, to the appendix regarding the actual sampling and storing conditions. The document is also intended to set an example for future documents and independent discussion forums on other kidney diseases for which emerging diagnostic and/or therapeutic strategies are reaching clinical practice.
Keywords: atypical hemolytic uremic syndrome, diagnosis, kidney transplantation, terminal complement blockage.
INTRODUCTION
Thrombotic microangiopathy (TMA), hemolytic uremic syndrome in particular, can be a devastating illness, with significant renal and non-renal morbidity and mortality. The awareness of the role played by complement in the pathophysiology of atypical hemolytic uremic syndrome (aHUS)1-3 and the introduction, in 2011, of a drug able to block the effects of complement system activation4 has dramatically changed the treatment and the prognosis of this entity.
However, its differential diagnosis is still challenging and this is at odds with the need for a fast diagnosis and early initiation of treatment in order to minimize organ damage. Despite the publication of several international consensus documents5-8, a recent survey involving European medical staff, mostly nephrologists with more than 10 years of clinical practice, revealed that approximately half of clinicians stated that making a diagnosis of TMA would take 3 days; similarly, treatment was delayed for at least 3 days in 57% of cases and in 13% of the cases for more than 1 week after presentation9.
In this same survey, only 70 and 78% of the enquired physicians requested complement evaluation and ADAMTS13 (A Disintegrin and Metalloprotease with ThromboSpondin type 1 motif, member 13) activity in the initial medical workup, respectively9. This reinforces the need to establish clear guidelines that may increase national and regional awareness of TMA, its differential diagnosis, and the proper lab test prioritization according to the local availability of these tests.
The Portuguese Society of Nephrology and the Society of Pediatric Nephrology of the Portuguese Society of Pediatrics working group on aHUS aims at establishing National recommendations for the differential diagnosis and management of aHUS by reviewing current definition and etiological classification of TMA, aHUS differential diagnosis and physiopathology as well an update on therapeutic management.
OVERVIEW OF THROMBOTIC MICROANGIOPATHIES
Thrombotic microangiopathy describes the pathological findings of microvascular endothelial injury and thrombosis. Originally introduced in 1952 by Symmers (10), it described the vascular lesions seen in Thrombotic Thrombocytopenic Purpura (TTP); later, Habib11 included under the same designation the pathological observations of HUS; since then, it has been recognized that these changes are present in many other conditions (e.g. scleroderma renal crisis, malignant hypertension) and its use has become more widespread11. In the heart of its pathophysiology lies an imbalance between the immune, clotting and complement systems, precipitated by environmental factors (prevalent in secondary TMAs) in the proper setting of genetic predisposition (prevalent in aHUS)12. Currently, this strictly pathological definition has evolved to a clinical diagnostic triad of a Coombs negative microangiopathic hemolytic anemia (MAHA), consumptive thrombocytopenia and platelet-mediated microvascular occlusion, leading to organ failure.
TMA classification can be both challenging and confusing. Ancient designations are not always reconcilable with the ever-evolving knowledge on pathogenesis.
Traditionally, HUS and TTP have been considered the two main variants of TMA. Around 90% of HUS cases were considered to be associated with infection by Shiga-Toxin producing Escherichia Coli (STEC), with these commonly designated as typical or diarrhea (D+) HUS7. The remaining non-STEC cases were referred to as atypical HUS13. Finally, the latter were either considered as secondary aHUS, whenever a cause could be identified, or, if not the case, primary aHUS13. More recently, alternative pathway complement dysregulation was found to underlie most aHUS cases and hence the designation of complement-mediated aHUS13. On the other hand, a deficit in the activity of the von Willebrand factor (VWF) cleaving protease ADAMTS13 is the hallmark of TTP; ultra large VWF multimers are formed, leading to platelet aggregation and microvascular thrombosis14. These definitions are reflected in the Kidney Disease: Improving Global Outcomes (KDIGO) consensus document of 20166. In our paper we will designate complement mediated HUS as aHUS. We refrained from using the designation of secondary aHUS for TMA conditions once STEC-HUS, TTP and complement mediated HUS are excluded and chose to designate these conditions as secondary TMA.
Upon presentation, it is critical to identify plausible causes as well precipitating factors, in order to initiate supportive or specific therapy within the first 24-48h after patient admission5. In figure 1 we present the general outline for the different diagnoses. Although the importance of a detailed clinical background (including family history and trigger events, such as infections and drug exposure) cannot be overemphasized, it is important to realize that the underlying cause is rarely identifiable on clinical grounds alone5. Hence the reliance on multiple laboratory tests; in Table 1 we present the key tests that should be ordered. We also reference Portuguese labs where these tests are available.
The key presenting findings that should evoke the differential diagnoses of TMA are:
– MAHA: low hemoglobin, elevated serum lactate dehydrogenase (LDH) level, undetectable (or markedly decrease) serum haptoglobin and the presence of schistocytes on a peripheral blood smear (although a non-obligatory criteria) with negative Coombs test.
– Thrombocytopenia;
– Organ injury (kidney disease, neurologic symptoms, gastrointestinal manifestations or other);
– Normal coagulation evaluation.
Etio-pathogenic classification of TMA
Atypical HUS
Atypical HUS, caused by dysregulation of the alternative complement, is essentially, but not entirely, a diagnosis of exclusion; i.e. mainly to be considered after ruling out TTP or STEC-HUS by testing ADAMTS13 activity and culturing for STEC, respectively5. But, and as detailed below, although driven by dysregulation of the alternative complement pathway, only a fraction of aHUS cases will display a decrease in serum complement components, C3 in particular, and in roughly half of them, mutations in genes encoding for the main regulatory proteins of the complement alternative pathway (or others) will not be identified. So, and although genotype and phenotype characterization of an aberrant alternative pathway complement are critically helpful for the diagnosis of aHUS, they will not be uniformly present in every single patient. In addition, even with the advent of the next generation sequencing technologies, genotype results can take several weeks1.
Treatment, therefore, has to be initiated based on a high degree of suspicion; not only excluding TTP and STEC-HUS, but being aware that secondary TMAs do exist and that they clinically and histopathologically may overlap with aHUS. A prompt diagnose of aHUS at presentation is one of the most challenging tasks a nephrologist can face these days. But is also one of the most rewarding. The major focus of the manuscript, aHUS is detailed in section III.
Thrombotic Thrombocytopenic Purpura
Thrombotic Thrombocytopenic Purpura (TTP) has a reported incidence of six cases per million per year in the UK and a mortality rate of 90%, that can be reduced by the prompt delivery of plasma exchange (PEX)15.
It is probably the main primary TMA to be considered in an adult patient. Though there is a lower incidence in children, we recommend the determination of the activity level of ADAMTS13 and, eventually, anti ADAMTS13 antibodies, in adolescents13,16. The key lab test is ADAMTS13 activity: levels < 5-10% confirm the diagnosis; a level > 5-10% rules it out15,17. It is mandatory to collect samples prior to initiating any kind of plasma therapy: donor plasma ADAMTS13 activity may confound results and plasmapheresis may remove patients anti ADAMTS13 antibodies, both instances leading to false-negative results. Although central nervous symptoms are key to TTP and renal involvement argue against it, one must realize that up to 35% of patients do not have neurological signs at presentation and that modest renal impairment can be present in up to 50% of the patients15. Platelet count can help in discrimination: usually between 10-30.000/μL for PTT while 50-100.000/μL for HUS15.
STEC-HUS
STEC-SHU is the major systemic complication of enterohemorrhagic Siga-Toxin producing Escherichia Coli infection, usually serotype O157:H7. Routine investigation for STEC-HUS is mandatory whenever diarrhea presents. Around 5% of STEC-HUS do not have a prodromal diarrhea and 30% of complement mediated
HUS do have diarrhea or gastroenteritis6. HUS complicates 6-9% of overall STEC infections and about 15% if children under age 10 are considered18. Around approximately two-thirds of STEC-HUS cases in the United States occurred among children less than five years of age19 and is the most common cause of pediatric HUS, accounting for 90 percent of cases20. But it also needs to be considered in adults: in the O104:H4 outbreak in Germany, 88% of the patients were 18 years of age or older21. Clinical manifestations may include history of bloody diarrhea in the previous 5-10 days, a peripheral white blood cell count above 10.000/μL and abdominal tenderness. Fever is frequently absent18.
Secondary forms of TMA
Beyond the diagnoses discussed above, several other entities may be associated with TMA. We generally define these as secondary TMA (see Figure 1).
One of the most common conditions associated with TMA that needs to be considered is severe hypertension. The differential diagnosis is sometimes difficult since TMA caused by hypertension is undistinguishable from all the other entities that cause TMA; also, most patients with other TMAs such as TTP or aHUS also present with new-onset severe hypertension. Hypertension causing MAHA and thrombocytopenia is typically severe, with systolic pressures over 200 mmHg and diastolic pressures over 100 mmHg, and is associated with severe kidney injury as well neurological symptoms related to posterior reversible encephalopathy syndrome22. The key to the differential diagnosis is the evolution of the clinical condition: TMA secondary to hypertension should improve quickly with blood pressure control, without the need for plasma therapy or terminal complement blockage17.
Systemic infections must also be considered while evaluating TMA. In the Oklahoma Registry, between 1989 and 2010, 31 (7%) of 415 patients were initially diagnosed as having TTP and therefore treated with PEX, but subsequently, the TMA was found to be secondary to a systemic infection17.
Streptococcus pneumoniae TMA needs to be excluded, particularly in the pediatric population, since 5% of TMA cases in children are associated with Pneumococcal invasive infections5. Children under 2 years have the highest incidence, similarly to other invasive pneumococcal infections23. Though invasive Streptococcus pneumoniae infections are common, these are only complicated by TMA in 0.4 – 0.6% of patients but are often more serious in terms of morbidity and mortality23.
The commonest precipitating illness for TMA is pneumococcal pneumonia with empyema (65–92%)24.
Pneumococcal meningitis has been less commonly reported though most of the patients with worse neurologic disease also presented with meningitis23. The clinical features can often cause confusion and delay in diagnosis due to a significant cross over with disseminated intravascular coaguIation25. Approximately 90% of patients have positive direct Coombs test results7. The epidemiology has changed with the emergence of different pneumococcal serotypes as newer pneumococcal vaccines became available. Pneumococcal 13-valent conjugate vaccine (Prevenar13®), included in our National Vaccination Program since 2015, is important to help prevent Streptococcus pneumonia TMA. However, Prevenar13® protects against many but not all HUS-related serotypes. Pneumococcal 23-valent conjugate vaccine covers all strains but is not recommended before 2 years of age. Although the pathogenesis of Streptococcus pneumoniae TMA remains uncertain, the Thomsen–Friedenreich antigen (T antigen) seems to play a central role23. Streptococcus pneumoniae produces neuraminidase, thereby exposing the T antigen on the surface of cell membranes, leading to IgM antibody activation via reaction with a complement-fixing antibody, resulting in red cell polyagglutination and hemolysis26. Ruling out Pneumococcal invasive infections is important since therapeutic plasma may aggravate the condition. Early identification of patients with Streptococcus pneumoniae TMA is critical so that fresh frozen plasma or plasma containing products for PEX may be avoided because most healthy donors will have anti–T IgM in their serum23,26. When Streptococcus pneumoniae infection is suspected or proven, it is recommended that washed blood products should be used23.
Autoimmune diseases can also be associated with TMA. The differential diagnosis may sometimes be difficult and there is a considerable overlap between entities12.
Indeed, mutations of the complement system may be present in up to 33% of the patients that present with HUS associated with autoimmune diseases12.
Clinically evident TMA is rare in Systemic Lupus Erythematosus (SLE), but occasionally superimposes in the anti-phospholipid syndrome and scleroderma.
In the case of SLE, TMA is mostly of histopathological nature. In a series of 149 SLE patients TMA was present in 36 (24.3%), but only 7 patients (4.7%) had associated
clinical features: in the remaining 29 patients TMA was exclusively a pathological finding in kidney biopsies, but one associated with severity and worse renal prognosis, as assessed by higher proteinuria, serum creatinine and total activity scores27. The pathogenesis of renal TMA in lupus nephritis remains unclear. SLE is an immune-complex mediated disease, so one can assume that the classical pathway drives TMA in lupus nephritis.
Nevertheless, several studies suggest that the dysregulation of the alternative complement pathway may play a role as well. For instances, nearly half of the patients with TMA features had decreased serum complement factor H levels and this was related to higher activity indexes and poorer renal prognosis27. Antiphospholipid syndrome (aPLS) can also manifest through TMA, particularly in patients with a catastrophic presentation, and successful use of eculizumab has been reported28. Finally Scleroderma Renal Crisis (SRC) may clinically and pathologically overlap with TMA, since, in addition to its clinical features of new onset severe hypertension and rising serum creatinine levels, MAHA is apparent in up to 50% of patients29. SRC is more likely to occur in patients with diffuse cutaneous systemic sclerosis, usually within the first 3–5 years of onset29. Contrary to SLE or aPLS, in SRC, complement dysregulation does not appear to be involved in the pathogenesis, and treatment rests on blood pressure control through blockage of the renin angiotensin system29.
Drug induced TMA may be mediated by drug-dependent antibodies reacting with platelets, neutrophils, and other cells, the best-documented case being quinine17.
Drugs may also cause TMA in a dose-dependent way17. Examples are mitomycin C, gemcitabine, calcineurin inhibitors (cyclosporine, tacrolimus [CNI]), mTOR inhibitors and vascular endothelial growth factor inhibitors (bevacizumab).
Disseminated malignancies may also be an occasional cause of TMA. Oncology literature series suggest that probably any metastatic malignancy may cause TMA 30. In most reports, the systemic micro vascular tumor emboli is the proposed mechanism of TMA17.
Clues that may suggest the presence of systemic malignancy include older age, weight loss, gradual onset of symptoms and peripheral leukoerythroblastosis17.
Among the cobalamin (Cbl) deficient inborn metabolism errors, the combined methylmalonic acidemia and homocystinuria (also termed CblC type), is the most frequent. Although an extremely rare cause of TMA, CblC should always be considered, particularly in the pediatric ages31-33. Failure to thrive, poor feeding, lethargy and neurologic abnormalities are usually present in infantile CblC-associated TMA31,32. However, late-onset in childhood and adulthood can occur. Particularly challenging is the late onset CblC presenting as isolated TMA in the absence of neurological involvement34.
Since serum total homocysteine determination is inexpensive and available in most hospital labs, we recommend that it should be performed at the initial workup of every TMA case, regardless of age or neurological involvement. If appropriate, the search for urine organic acids, serum methylmalonic acid (MMA) and plasma amino acids (low methionine levels) should follow. Molecular diagnosis can be accomplished by MMACHC gene sequencing. In Portugal, the MMACHC c.271dupA (p.Arg91Lysfs*14) allele is the most prevalent, either in homozygosity or compound heterozygosity35.
The treatment goal is to normalize serum methionine and to lower homocysteine and MMA as soon as possible, which can be achieved through the administration of hydroxocobalamin and betaine31,32.
Pregnancy is associated with various forms of TMA, accruing significant perinatal and maternal morbidity and mortality36. The differential diagnosis of TMA in pregnancy primarily concerns aHUS and TTP, as well as the TMA occurring in the syndrome of hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome)37, which is part of the clinical spectrum of preeclampsia38.
TMA occurring late in the third trimester or postpartum is usually caused by aHUS39-41. A high proportion (>50%) of these women will have identifiablecomplement mutations, and pregnancy acts as a trigger in those with an underlying genetic predisposition42.
This provides the rationale for complement inhibition therapy, and increasing data suggests that eculizumab is safe during pregnancy43,44. According to KDIGO recommendations all patients with pregnancy-associated HUS should have a full complement evaluation.
On the other hand, TMA that occur during pregnancy, particularly in the second and third trimesters and resolve following pregnancy, are usually TTP.
This might be explained by the physiologic increase in von Willebrand factor during pregnancy, which consumes ADAMTS1342. Accordingly, on the basis of observational data and knowledge of pathogenesis, PEX is recommended45.
Although the etiology of HELLP and preeclampsia is not fully understood, it has been linked to elevated circulating antiangiogenic factors soluble Flt1 (sFlt1) and endogline. sFlt 1 reduces the concentration and activity of vascular endothelial growth factor (VEGF), leading to endothelial dysfunction, hypertension, and proteinuria37. There is some evidence that complement is activated in preeclampsia and HELLP, although it is unclear whether it plays a role in pathogenesis, and seems to be one of many predisposing factors rather than the main triggering event42 in contrast to pregnancy-associated aHUS. Management should be supportive, and the definitive treatment of HELLP is expedited delivery.
Histopathological Features of Thrombotic Microangiopathies
Endothelial cells are crucial players in TMA. Their damage is the most important event in the pathogenesis of the disease and lesions are predominant in vessels, consisting in microthrombi with red blood cells fragmentation in arterioles, capillaries and arteries46.
It is also important to realize that severity and duration of the disease do influence the development of these features, either in glomeruli or vessels. The endothelial cell aggression commands almost all the morphological aspects in the early stages of the disease.
Light microscopy findings in glomerular early lesion consists of endotheliosis, the swelling of the glomerular endothelium, with subendothelial widening that, depending of the intensity of the aggression, may occlude the capillary lumina. Eventually, bloodless glomeruli (closure of capillary lumina) may be observed ( Figure 2a). The separation of the endothelial cell from the basement membrane with production of a new basement membrane gives rise to double contours, best observed by PAS or Silver stains. Congested glomeruli, or glomerular paralysis, is due to the presence of red blood cells in the glomeruli, in cases where there is severe involvement of the efferent arteriole. Fibrin can be detected within thrombus or in cases where there is fibrinoid necrosis of the afferent arteriole. Exceptionally, crescents may develop in some cases, secondarily to necrosis.
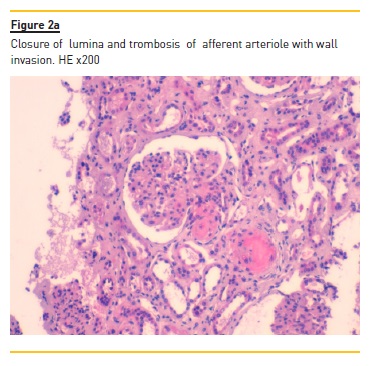
Additional findings include glomerular capillary infiltration by large number of neutrophils, a mesangial fibrillary appearance due to edema and mesangiolisis, the dissolution of mesangial matrix. The glomeruli in biopsies performed later along the natural history of the disease (Figure 2b) will display a solid aspect, reflecting a collapse of the glomerular tuft, with thickening and wrinkling of capillary walls, as a consequence of severe vascular lesions and associated ischemia. The migration of mesangial cells to the sub-endothelial space will consolidate the double contour pattern, sometimes mimicking a membranoproliferative aspect.
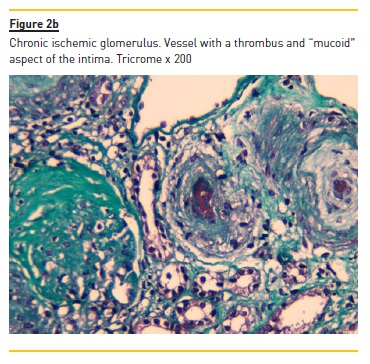
Also, a continuous augmentation of matrix due to ischemia will lead to mesangial sclerosis. In far advanced disease, the ischemic aspects of glomeruli are more frequent: in the chronic ischemic glomeruli there is a simplification of the glomerular tuft, which comes smaller, with the resulting widening of the Bowman space being filled by collagen.
Various changes may occur in arteries and arterioles in TMA. Some cases have mild aggression to the vessels, but whenever it happens, it is the severity of the lesions of vessels that dictates prognosis. In arterioles, once again, endotheliosis induces narrowing of the capillary lumen, thrombi may develop, fibrin may invade the arteriolar wall and necrosis tend to occur at the hilum of the glomerulus. In arteries, swelling of the intima is responsible for the intimal mucoid appearance, thrombi are present, as is fibrin in the lumen, sometimes invading the wall. Infiltration of the intima by cells may induce a pattern of onion skin, the hallmark of evolution towards fibrosis11.
Immunofluorescence findings tend to be scarce and inconstant. The majority of biopsies will not display deposits. Fibrin in thrombi or areas of fibrinoid necrosis of glomerular capillaries and vessels is expected. IgM may be seen in parietal vessel walls. In some cases, C3 or C5b-C9 have been reported in glomeruli, not only in aHUS but also in TMAs associated with drug toxicity or after hematopoietic stem cell transplantation. It is striking why complement is so rarely seen in aHUS biopsies, a disease driven by the alternative pathway dysregulation. There are two hypotheses to justify this point: one is technical; the actual immunofluorescence method may not be sufficiently sensitive to label them; another hypothesis relates to the time lag between the acute phase of the disease and time of biopsy, usually a few weeks after presentation and once treatment-induced hematological remission has occurred: by then, complement components may have already been degraded6,11,47.
Ultrastructural studies of biopsy specimen are rather non-specific and affect mainly the endothelial cell, with sub-endothelial expansion, swelling and loss of fenestration.
Similarly to what was already stated for the clinical findings, the reported morphological aspects are shared among TMAs in general, and cannot be used to differentiate them. However, for an experience nephropathologist, subtle changes may suggest a specific diagnosis. For instance, in aHUS the thrombi are richer in fibrin, whereas in TTP the thrombi are full of platelets.
Renal biopsy remains an important procedure in the evaluation of TMA. As abovementioned, per se it will not establish the etiology of any given TMA, nor is it, with the current awareness of TMA (aHUS in particular), expected to provide the first clue to a TMA or aHUS diagnosis, with the occasional exception of TMA occurring in renal allografts. Still, it is critically important in ruling out secondary forms, such as TMA in lupus nephritis, and establishing a reliable renal prognosis.
ATYPICAL HUS
General Overview: Epidemiology, Clinical Presentation and Physiopathology
Atypical hemolytic uremic syndrome is an ultra-rare disease. In Portugal, the true incidence and prevalence is unknown. In Europe aHUS has an estimated prevalence of 2/1,000,000 adults and 3/1,000,000 children.
This disease can manifest in any age, but usually affects predominantly children and young adults. Gender distribution appears to be similar48,49.
The diagnosis of aHUS is mainly clinical, because as previously mentioned, complement abnormalities are not uniformly present and secondary TMA forms are essentially ruled out on clinical grounds alone. Therefore, the diagnosis is based on the combination of clinical evaluation and biochemical findings. Additionally, results of genetic testing are not immediately available.
The classical presentation is laboratory evidence of the classical triad of hemolytic anemia (decreased hemoglobin, elevated LDH, decreased haptoglobin, schistocytes on blood smear) with Coombs test negative, thrombocytopenia and acute kidney injury. However, a subacute presentation can occur with proteinuria, acute kidney injury, arterial hypertension with signs of TMA on renal biopsy with or without thrombocytopenia and hemolysis. Therefore, in every patient presenting with renal insufficiency and low-grade hemolysis the differential diagnosis with TMA needs to be considered50.
Extra-renal findings are present in up to 20% of the patients. Gastrointestinal involvement (diarrhea, colitis, abdominal pain, and pancreatitis) was documented in one-fourth of aHUS patients. Also, some of these patients have bloody diarrhea. Neurological symptoms were also found, as cardiac (acute myocardial infarction, cardiomyopathy, heart failure) and pulmonary49,51.
The complement system (Figure 3) is an essential part of innate immunity and provides a first-line defense against invading pathogens infections and non microbial forms of stress. Three distinct activation pathways belong to the complement system: the classical, lectin, and alternative pathways, all resulting in the formation of C3 convertases. The C3 convertases (C4b2a and C3bBb) continuously cleaves C3 in an amplification loop. The terminal complement cascade is initiated by the C5 convertase and generates the membrane attack complex inducing cell lysis. The C3 convertase amplification loop requires rigorous control to prevent unintended tissue inflammation and damage52.
In aHUS, the underlying pathogenesis lies behind the alternative pathway dysregulation on host cell surface, secondary to complement gene mutations or presence of antibodies against complement factors.
The dysregulation of the alternative complement pathway can result from either a loss-of-function mutation in a regulatory gene (CFH, CFI, MCP and THBD [thrombomodulin coding gene]) or a gain-of-function mutation in an effector gene (CFB and C3). Penetrance of aHUS in carriers of mutations in some genes is around 50%53-57.
Also, the presence of autoantibodies against factor H (fH)58,59 that interfere with the alternative pathway regulation in a similar way to mutations, has been reported in aHUS children. Usually the presence of antibodies against (anti-fH) is associated with a homozygous deletion of the genes for complement factor H-related (CFHR) proteins (CFRH1 and CFRH3)60.
In addition to the mutations in complement proteins, some patients with aHUs present mutations in other molecules not directly linked to complement system, such as mutations in diacylglycerol kinase ε, plasminogen, and factor XII (although only in the presence of anti-factor H autoantibodies). Some patients have mutations in the thrombomodulin coding gene, which has a role in both coagulation and complement regulation61,62.
Diagnostic of atypical Hemolytic Uremic Syndrome
General evaluation
The diagnosis of aHUS relies, firstly, on the proper characterization TMA as above mentioned (section II.a).
Kidney injury is identified as creatinine elevation and hematuria or proteinuria. Figure 1 summarizes the diagnostic work-up to perform in these patients.
As a priority, TTP needs to be ruled out. An ADAMTS13 activity <10% confirms the diagnosis of TTP (see Table 1 for sample conditions). Excluding STEC-HUS comes next. Screening specifically for E. coli O157:H7 serotype in stool with sorbitol-MacConkey (SMAC) agar is often considered. However, other serotypes of STEC have been reported to account for a significant fraction of outbreaks21. Therefore, whenever diarrhea is present, the direct detection in stool specimens of one or two phage-encoded Shiga toxins, Stx1 and Stx2, by an enzyme-linked immunosorbent assays (ELISA) is preferred as the initial approach. Once stool cultures are positive and available, detection of the responsible genes, stx1 and stx2, by PCR amplification techniques (real-time PCR) using commercial kits, is another possibility, depending on local availability. In the event of a highly suggestive STEC-SHU case but with negative ELISA and/or real-time PCR assays for the Shiga toxins, bacteria isolated from stool cultures should be referred to the Instituto Nacional de Saúde Doutor Ricardo Jorge, Lisboa, for further molecular bacterial DNA genotyping63.
After the exclusion of these two conditions, secondary causes of TMA, which will eventually require specific treatment, need also to be ruled out (see Figure 1).
Phenotype evaluation of the complement
The levels of complement proteins should be evaluated in all patients with TMA and clinical suspicion of aHUS, prior to plasma therapy6. In Table 1 we present the complement studies which must be carried out in these patients.
It should be noticed that in general, low C3 and normal C4 concentrations in serum suggests complement alternative pathway activation. But one must be aware that decreased serum C3 level is not specific for aHUS and also that normal C3 and C4 concentrations do not exclude the diagnosis of aHUS47.
A large decrease in C3 is usually observed with mutations in C3, CFI and CFH genes. Factor B (fB) is a protein unique to alternative pathway, and a decrease in levels of fB is an indication of activation of alterative pathway.
Factor I and fH should also be evaluated, since low levels of these tend be associated with low C3 levels.
Quantification of MCP factor (CD46), found to be reduced in 10% of patients with aHUS, can be performed by flow cytometry and, importantly, the levels of MCP factor are not influenced by plasma therapy47. Recent guidelines recommend the evaluation of CD46 surface expression by flow cytometry6 but, in Portugal, no lab is currently performing this test.
Many complement functional assays and activation markers were, in the meantime, developed. CH50 is the most used assay to screen complement abnormalities.
A low CH50 can result from congenital complement deficiencies, increased consumption of complement factors or insufficient syntheses of complement factors. AH50 is used to evaluate the alternative pathway activity. If the test is performed during the acute phase of the disease, the AH50 activity is low because of consumption of complement cascade components47. The KDIGO recommendations are to evaluate CH50 and AH50 in all patients with aHUS suspicion6.
Additionally, complement activation markers are being evaluated, including C3 decay products (C3a, C3b), C4 decay products (C4d), C5 decay products (C5a) and fB decay products (Ba, Bb). The most interesting of these products is the fB decay products, in particular Bb. The fB is specific for alternative pathway and elevated levels of Bb fragment with decreased levels of fB were reported in patients with aHUS(47, 64).
However, the results obtained with complement activation markers are conflicting and not recommended in evaluation of these patients, according to international guidelines6. These tests are not available in Portugal and there is no evidence to recommend its use.
Human molecular genetics of the complement
Atypical HUS is characterized by dysregulation of the alternative complement pathway resulting from either a loss-of-function mutation in a regulatory gene (CFH, CFI, MCP or THBD) or a gain-of-function mutation in an effector gene (CFB or C3)(65, 66). The mutations were mainly found in the heterozygous state, and approximately 5% of patients have combined mutations, usually in CFH with either CD46 or CFI. Homozygosity for risk haplotypes of CFH (rs3753394, c.1-332C>T and rs1065489, c.2808G>T, p.Glu936Asp) that tag the disease risk haplotype CFH-H3 and one polymorphism in MCP (rs7144, c.*897T>C) that tags the MCPggaac risk haplotype have been shown to significantly increase disease penetrance and severity(67-69).
Additional genetic risk factors include a deficiency of CFH–related proteins 1 (CFHR1) and 3 (CFHR3), due to CFHR1–3 deletion in homozygous state caused by non-allelic homologous recombination of CFHR3 and CFHR1(70, 71). Finally, recessive mutations in DGKE, which encodes diacylglycerol kinase-ε and is expressed in endothelial cells, platelets, and podocytes, were identified in children with the onset of aHUS in the first year of life16.
However, detectable complement abnormalities have been described in only approximately 50%–60% of patients; therefore, the aHUS diagnosis is based on clinical criteria and the exclusion of a severe ADAMTS13 deficiency (<10%) and STEC-HUS72.
The advent of next-generation sequencing (NGS) allows simultaneously sequencing of large gene panels and generates competitive results at a lower cost and in a shorter amount of time.
Therefore, the strategy for TMA diagnosis should be based on a workflow that includes the measure of ADAMTS13 activity and screening of ADAMTS13 and complement genes using a NGS gene panel – ADAMTS13, CFH, CFHR1, CFHR3, CFHR4, CFHR5, CFI, CFB, C3, THBD and DGKE (see Annex 1 – Human molecular genetics: information for sampling and where to send and Annex 2 – Informed consent for genetic testing).
The NGS-targeted gene panels have changed the paradigm of routine molecular studies. In the face of the multiple genetic changes found in every patient, the critical challenge was discriminating disease-associated variants (historically referred as mutations) from the broader background of variants present in all patients genomes(73-78). The prediction of pathogenicity of the genetic variation has become crucial for understanding the great inter-individual variability of these patients.
According to the practice guidelines for the evaluation of pathogenicity recently published by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology77 the following criteria must be evaluated: 1) whether the variant was a stop/frameshift variant, which was considered to most likely be disease causing, 2) co-segregation in the family, 3) whether the variation had been previously identified in international databases, 4) in silico evaluation and 5) presence of the second mutant allele in the case of autosomal recessive inheritance. Thus, the variants will be classified as pathogenic, likely pathogenic, uncertain significance, likely benign or benign based on the available evidence.
This approach provides an accurate molecular analysis; however, its interplay with a detailed clinical data registry sustained by a multidisciplinary team is crucial for a correct TMA differential diagnosis (see Annex 3 – Clinical information).
Outcome
The availability of eculizumab has significantly modified the prognosis for patients with aHUS. With PEX alone, the mortality was about 10% in the first episode and more than 50% of the patients needed dialysis during the first year of the disease48,49.
Clinical outcomes remain critically dependent on the genotype. Table 2 summarizes patient survival as well both native and graft kidney survivals according to genotype, based on which we can stratify the risk of death or End-Stage Renal Disease (ESRD), recurrence and transplant relapse. This information was compiled from several published aHUS cohorts48,49,69. Genophenotype correlations in a cohort of 273 aHUS patients revealed49 that complete remissions were more common with THBD and MCP mutations (62% and 90% respectively), and that patients displaying MCP mutations also spontaneously remitted more frequently.
Poor responses were observed in patients with CFH and CFI mutations, with a complete remission rate of only 5% and 12.5%, respectively. Additionally, partial remission was observed in more than 50% of treated patients with CFH mutations or in the presence of antifH antibodies. Overall, better treatment responses were observed in children than adults (78% vs 53%) and mortality or ESRD after plasma therapy was high, especially with CFH (77%) and CFI (67%) mutations49. In another retrospective analysis of a cohort of 214 patients with aHUS, more than half of adult patients with CFH mutations developed ESRD after the first episode, despite treatment with high volume PEX48.
Similarly, the outcome of kidney transplantation is influenced by genetics: 60% of patients had recurrent disease, with an 80–90% of graft loss, depending on the causative mutation(79, 80). Once again, mutations in CFH are associated with a higher risk of recurrence or graft loss after transplantation (75-90%), particularly those related to abnormalities in terminal 3 or gene conversions between CFH and CFHR1 (hybrid gene CFH/CFHR1)81. In two French case series(51, 82) the rate of post-transplant aHUS recurrence in patients with CFH mutations was around 75–80% in 5 children and 16 adults who received 6 and 17 renal transplants, respectively.
The same recurrence rate was noted in a 2006 meta-analysis of 36 renal transplantations in 27 patients with aHUS associated with CFH mutations, leading to a graft loss in 93% of patients79. In aHUS patients with CFI mutations, the post-transplant recurrence rate is also high and associated with poor prognosis56,83,84.
More than half of cases with mutations in CFI had an additional genetic susceptibility for aHUS, which negatively impacted kidney survival85. Patients with MCP mutations are expected to have a low risk of post-transplant aHUS recurrence (<20%) because MCP is a membrane-bound protein, expressed on endothelial cells and, therefore, transplantation will restore normal renal endothelial MCP function83. However, a recent large study69 revealed an unexpected recurrence of disease after transplantation in some carriers of MCP mutations. This is probably explained by the fact that a significant proportion of patients with mutations in MCP also carried mutations in genes encoding circulating complement factors69. Because mutations in other genes are rare, less data is available about the risk of recurrent disease. Nevertheless, existing evidence suggests significant risk of recurrent associated with mutations in C3 and CFB, 60% and 100%, respectively, with a high risk of graft failure on recurrence(79, 86).
The risk of post-transplant recurrence in patients with THBD mutations is not well-established, because thrombomodulin exists both in membrane bound and soluble forms, hence, it is not possible to reliably predict the risk of post-transplant recurrence in carriers of these mutations. Considering the available studies(87-89) THBD mutations may favor recurrence after kidney transplantation. Concerning patients with anti-fH antibodies, the risk of post-transplant aHUS recurrence seems to be related to high and persistent titers of antibodies(59, 82). No posttransplantation recurrence has been observed to date in patients with DGKE mutations16.
Finally, data on the rate of recurrent aHUS after transplant in patients with no identifiable pathogenic alleles is sparse. Overall, the risk seems to be lower (30%)82.Therefore these patients have to be considered at medium risk of recurrence.
Treatment of atypical HUS
Administration of eculizumab is recommended as first line treatment in paediatric patients with suspected aHUS. Taking into account the technical difficulties of PEX in paediatric patients and potential complications, in addition to the superiority of eculizumab for the recovery of renal function, early first-line treatment with eculizumab is recommended in this population and the use of plasma exchange should be completely avoided.
For adult patients, in the presence of an unequivocal diagnosis of aHUS we recommend the use of eculizumab as first line therapy. Patients presenting with a TMA of uncertain etiological diagnosis, we recommend starting PEX. Once secondary causes of TMA, STEC-TMA and PTT have been excluded, a presumptive diagnosis of aHUS can be assumed in these cases, and if the patient remains plasma exchange dependent or displays plasma resistance, eculizumab is to be initiated.
Plasma therapies in aHUS
Both plasma exchange and plasma infusion (PI), by the administration of large volumes of fresh frozen plasma, can provide functional complement-regulating proteins. PEX has the advantage of removing dysfunctional complement factors, as well as anti-fH antibodies and potential inflammatory molecules that might result from endothelial injury, while reducing the risk of volume overload5,90-92.
The use of PEX in the setting of aHUS was attempted for the first time in 197993. In the next years, other case reports and small series of patients successfully managed with PEX have been described90. However, some of those reports included TMA secondary to other conditions. Furthermore, the role of complement in the pathogenesis of aHUS was not yet established, consequently no genetic information was available, and we can only assume the presence of aHUS based on the exclusion of other known causes of TMA. Still, some degree of improvement was observed in about 87% of the patients in a disease with a previous high morbidity and mortality rates90. Over the next few years, early and intensive, with high volume PEX was empirically used in the treatment of aHUS92,94,95. Potential complications associated with PI and PEX are more common in children than in adults and include hypotension, allergic reactions, nausea and vomiting, hypocalcaemia and catheter-related complications96,97.
Though actual recommendations advocate first line therapy with eculizumab particularly in children5,6,16, PEX still has its role in the management of aHUS patients.
Eculizumab is not available (or promptly available) in every center. When a delay in eculizumab administration is expected, PEX should be initiated within the first 24 hours, considering the poor prognosis associated with late treatment6,16,94. Patients with complete hematological and renal recovery after PEX may not require switching to eculizumab5,16. PEX is also recommended in the presence of anti-fH antibodies in combination with immunosuppression given the favorable outcome on renal function and mortality16,91,98-102. In the presence of severe disease, with coma or seizures, and while TTP is not yet ruled out by the presence of ADMTS13 activity > 10%, PEX should also be considered6.
When PEX is the selected therapy option, the replacement fluid should be fresh frozen plasma in order to provide functional complement factors and the volume should be 1.5 of plasma volume (60-75ml/Kg). Different treatment schedules have been reported in the literature and cannot be compared. It is recommended to maintain treatments until normal platelet count is obtained, hemolysis stops and sustained improvement in renal function is observed. Protocols of 5 daily treatments initially, followed by 5 treatments per week for 2 weeks and then 3 treatments per week for the next 2 weeks have been proposed. Subsequent treatment should be assessed on individual basis5,16.
Terminal complement blockage
The complement system offers multiple potential therapeutic targets, and several drugs acting on the activation pathways, the anaphylatoxins, the amplification loop and the terminal pathway, have been developed and entered preclinical and clinical trials103.
Blocking the terminal pathway offers the possibility of having a potent inhibitory effect on the cascade, while at the same time allowing the upstream components to retain some of their physiological functions, such as the removal of apoptotic and necrotic cells and solubilization of immune-complexes104.
Eculizumab4 is a humanized monoclonal IgG2/4 kappa antibody against C5 that blocks cleavage of the terminal complement protein C5 into the proinflammatory C5a and lytic C5b-9. After being developed in the 1990s, it was tested in a range of inflammatory conditions in early clinical trials. The most impressive efficacy was found for Paroxysmal Nocturnal Hemoglobinuria (PNH), and it was for this condition that it was firstly approved for clinical use in 2007 by the FDA (Food and Drug Administration) and EMA (European Medical Agency). As another prototypical disease resulting from complement dysregulation, aHUS was the next obvious candidate for clinical use of eculizumab. After successful case reports105,106, and the preliminary reports of two prospective clinical trials107,108 of primarily adult patients, either with evidence of progressing TMA or with long disease duration, demonstrating efficacy and safety of eculizumab in aHUS, it became in 2011, the first and only approved treatment for aHUS.
In 2016, the first and largest study of eculizumab to treat severe aHUS in an exclusively adult population was published. An open-label single-arm phase 2 trial, with 41 aHUS adult patients receiving eculizumab, highlighted its benefits in improving hematological, renal and quality of life paramaters, dialysis discontinuation and transplant protection, after a 26-week treatment period109.
Although there has never been a randomized controlled trial on eculizumab in aHUS, the mechanistic rationale and the high morbidity and mortality associated to this disease, justified that comparison with historical controls is sufficient enough for the recommendation of its use. And clearly, the prognosis of aHUS has been transformed: full recovery of renal function is now expected, other than in those who present late in the course of disease. KDIGO guidelines recommends that all patients with a clinical diagnosis of aHUS are eligible for treatment with a complement inhibitor6.
The recommended dosing schedule for eculizumab in aHUS is the one reported in the trials (Annex 4 – Eculizumab dosing schedule), and although options for altered dosing have been considered such as the minimal dose required to achieve complement blockade or a discontinuation dosing schedule, there is still not enough data to support either option108,110.
In patients with ant-fH mediated aHUS, eculizumab should be used in patients with severe injury of vital organs16, in association with immunosuppression. Eculizumab discontinuation should be guided by antibodies titers6,16.
Prophylaxis of meningococcal infections
All patients should be immunized against meningococcal infection (including type B) given the risk inherent to the treatment with eculizumab. Ideally, the vaccination should be given at least two weeks before treatment or getting clearance for transplantation.
However, eculizumab initiation should not be delayed because lack of previous immunization and this circumstance antibiotic prophylaxis is mandatory up two weeks after vaccination6. Currently, in Portugal, the following vaccines are available with the preconized schedules for the general population111:
a) Conjugated tetravalent meningococcal vaccine – MenACW135Y (Nimenrix®, Menveo®)
b) Meningococcal B vaccine – MenB (Bexsero®, Trumemba®)
In addition to invasive meningococcal disease, treated individuals are at increased risk of serious infection by other capsulated bacteria namely Haemophilus influenza type b and Streptococcus pneumoniae.
Patients vaccination status must be confirmed in accordance with the recommendations of the Direção Geral da Saúde (DGS)112.
The working group therefore recommends the following scheme:
– Meningococcal vaccination (mandatory): Men-ACW135Y and MenB
– Others (recommended): Pn13, Pn23, Hib, Influenza Since the carriage rate of meningococci is the highest in adolescents and young adults (up to 30% between age 16 and 24) vaccination of susceptible household close contacts may be also considerer (siblings and parents)16.
Antibiotic prophylaxis
The current vaccination scheme is not effective in protecting against all meningococcal serotypes. Also, it is unknown if anti-meningococcal antibodies are protective in the setting of complement blockade. Therefore, giving prophylactic antibiotic treatment to all patients under eculizumab therapy is recommended113.
Prophylactic treatment for adults and children > 12 years old is to be performed with amoxicillin 500 mg orally twice daily; for children two-12 years of age, 250 mg orally twice daily; and < two years old 10 mg/kg twice daily. In the presence of penicillin allergy, a macrolide can be used: for children and adolescents, we recommended azithromycin at a dose of 5 mg/kg/day. The antibiotic prophylaxis should be used during throughout the treatment and until 60 days after eculizumab treatment has ended6.
Neither vaccines nor antibiotic prophylaxis guarantee full protection114, hence the importance of patient/family educations about signs of meningococcal infection and the availability of an information card to be carried by patients or their care-giver16.
Pregnancy
Recent studies, including those on PNH patients, showed acceptable outcomes with a high rate of fetal survival and a low rate of maternal complications, although preeclampsia and HELLP syndrome have not been prevented in 2 and 1 reported cases respectively, and higher doses of eculizumab than generally recommended had to be used during pregnancy43,44.
Monitoring disease activity and therapeutic efficacy
Complement blockade is obtained within 1h of the first dose. With the recommended treatment scheme, most patients will have full complement blockade in between consecutive doses. Special attention should be paid to children and pregnant women who could need an adjustment to the usual dose5,16. Also, patients with massive proteinuria may need higher doses of eculizumab.
During the treatment, monitoring relies on regular measurement of CH506, consisting in the use of activated sheep erythrocytes which are mixed with dilutions of patients serum to identify the dilution required to lyse 50% of available sheep erythrocytes.
Eculizumab treatment suppresses CH50 activity, so in these patients is expected to obtain a CH50 < 10 % of normal value, using this technique47,115. Easier hemolytic or enzyme-linked immunosorbent assays are currently commercially available, and normal range is assay dependent. A limitation of this test is that CH50 cannot be used in patients with complete fH deficiency (e.g. of homozygous CFH mutation) in whom CH50 levels are permanently undetectable regardless treatment status115.
Eculizumab trough levels could be measured in patients under this treatment and is expected to have full complement blockade with trough levels above > 100 μg/mL6,16. In Portugal, no lab yet performs this technique.
Eculizumab discontinuation
One of the most controversial issues concerning the use of eculizumab in aHUS, is the duration of treatment.
Some current recommendations suggest that eculizumab should be prescribed indefinitely5. Supporting this notion is the fact that aHUS is a chronic disease and TMA manifestations are unpredictable and can lead to irreversible and potentially life-threatening complications116.
On the other hand, continued therapy with eculizumab carries an increased risk of meningococcal infection, the need for prolonged prophylactic antibiotherapy and is very costly. In fact the optimal duration of eculizumab treatment is presently unknown6. In the original eculizumab trials108 a continued and time dependent improvement of kidney function was observed up to 12 months (quick increase in estimated glomerular filtration rate (eGFR) in the first 26 weeks and a trend towards stabilisation thereafter). In one of the trials (trial 1) 4 out of 5 patients that were dialysis dependent recovered autonomous kidney function.
Based on these observations we follow other consensus boards5,6,117 and recommend that eculizumab should be maintained for a minimum of 6-12 months; also, in patients with acute renal failure in need of renal replacement therapy, eculizumab treatment is recommended for at least 3 months before establishing the final diagnosis of end stage renal disease.
If suspension of eculizumab therapy is being considered, the risk of relapse should be discussed with the patient. A recent analysis of discontinuation of eculizumab therapy in patients with aHUS revealed that treatment was discontinued in 47% of the patients included in the original clinical trials and in 26% of the patients included in aHUS registries116. Relapse rates ranged from 20% to 31% and in 5%, organ losses were reported116. This analysis was unable to establish any definitive predictor of relapse, although patients with relapses seemed to have a higher proportion of CFH gene pathogenic variants116. The authors consider that until novel tools enabling a more robust risk stratification and adequate monitoring of complement activation and disease activity are made available, the option to discontinue eculizumab therapy will not be an evidence-based decision116.
However the analysis of the French aHUS Registry, including 108 patients, identified 38 in which eculizumab was suspended (35%); of these, 12 (32%) relapsed with a median time to relapse of 7,5 months (total follow up time was 22 months)109. Of the 12 patients that relapsed, 8 (67%) had CFH gene pathogenic variants (compared with only 3 nonrelapsers with CFH pathogenic variants, p=0.002) and 4 (33%) had MCP pathogenic variants (compared with 4 nonrelapsers with MCP pathogenic variants, p=0,2). Importantly, all patients that relapsed had an identified pathogenic variant in one of the complement genes (p=<0.001)109.
A key aspect of this analysis was that eculizumab was resumed in all relapsing patients within 48 hours, leading to a good therapeutic response: within the follow-up period no changes in eGFR or relapses were reported109.
In conclusion, the optimal duration of eculizumab treatment in patients with aHUS is currently unknown.
If suspension is being considered, a risk analysis of relapse should be made. Presently, the best available data supporting any decision resides on the published analysis of the French aHUS Registry: in patients with no mutations or MCP mutations eculizumab discontinuation can be considered; in patients with CFH pathogenic variants any decision must take in consideration the high risk of relapse; and in patients with anti-fH antibodies, eculizumab discontinuation can be considered only when titers have been significantly reduced109.
In all cases of discontinuation of therapy patients must be followed closely. Regular blood and urine tests, including serum creatinine, haemoglobin, platelet count, schizocytes, lactate dehydrogenase, haptoglobin and proteinuria-to-creatinuria ratio, should be performed every week for first month, subsequently every two weeks for 3 months and monthly thereafter109; regular blood pressure monitoring is also suggested.
Patients should consult their physician in case of poorly controlled blood pressure or any intercurrence that might trigger the disease (e.g. infection, surgery, pregnancy).
Importantly, in case of a relapse, eculizumab therapy should be quickly resumed.
Immunosuppressive therapy
The use of immunosuppression has shown benefit in children with anti-fH antibodies. Steroids in combination with cyclophosphamide or rituximab following PEX reduce antibody titers, prevent disease relapses and improve renal survival(69, 85-87). In a cohort of 138 Indian children with anti-fH antibodies, early combined therapy with PEX and induction immunosuppression was significantly associated with better outcomes in the multivariate analysis (OR 0,22, 95% CI 0,07–0,76; P = 0,016). One case report of an adult successfully treated with PEX followed by steroids and rituximab has also been described88.
Kidney transplantation
Kidney transplantation should be delayed until at least 6 to 12 months after the start of dialysis because renal recovery may occur several months after starting eculizumab118,119. The resolution of hematological and other extrarenal TMA manifestations is a prerequisite for transplantation.
Recurrence of aHUS following renal transplantation
Post-transplant aHUS recurrence is usually an early event, with 60% of cases occurring within the first month after transplantation79. This higher risk of early recurrence (Figure 4) may relate to endothelial activation by ischemia-reperfusion injury, drugs (e.g. CNI and mTOR inhibitors), infections and alloimmune responses (e.g. donor specific antibodies). Although early recurrence is most frequently seen, late recurrence, up-to several years after transplantation, has also been reported120.
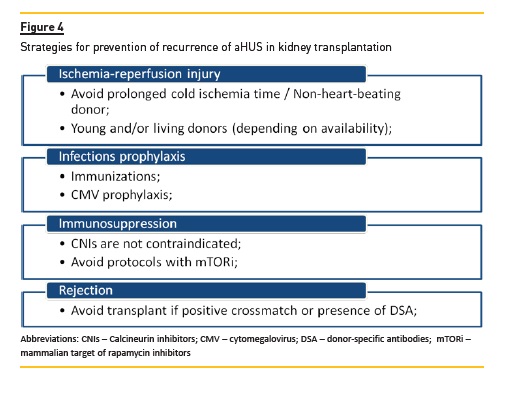
More important, the risk for posttransplantation recurrence is dictated by the underlying genetic defects (see above, section c. Outcome) and the recurrence in previous grafts117. According to this information, the risk of recurrence of aHUS can be stratified for patients who are candidates for renal transplantation (Table 3).
De novo TMA following renal transplantation
De novo post-transplant TMA (Figure 5) can develop in the absence of previous aHUS or any known susceptibility factor for aHUS. However, approximately 30% of patients who develop de novo post-transplant TMA carry mutations in complement regulatory proteins encoding genes82.
Post-transplant immunosuppression regimens
Several strategies related to immunosuppression regimens have been attempted to prevent recurrence of aHUS in the kidney grafts.
Treatment with CNI, cyclosporine (CsA) or tacrolimus, are reported to be associated with post-transplant TMA121,122, the risk being apparently higher for CsA (up to 14% of patients in one series)121. The use of mTOR inhibitors bears a greater risk for post-transplant TMA and it was shown that early use of mTOR inhibitors is an independent risk factor for the development of TMA123, with CNI having a synergic effect124.
Data on the post-transplant immunosuppressive regimen and the risk of recurrent aHUS is controversial.
A small study suggested that the early use of CsA increased the risk of aHUS recurrent in adults125, but analysis of a larger pediatric cohort failed to confirm it126. Therefore, avoidance of CNI treatment is generally not believed to reduce the risk of recurrent aHUS79.
Immunosuppression based on belatacept could be followed depending on the immunological risk of each patient, although, until now, no conclusive data are available in the literature.
Living kidney donation
Living related donor transplantation has traditionally been contraindicated for patients with aHUS, because it associates with a risk for recurrence in the recipient and de novo disease in the donor, if the former carries an at-risk genetic variant127,128. For the same reason, any potential donor with evidence of complement dysregulation should be excluded.
However, if an at-risk genetic variant is detected in the patient, it will be possible to consider living related donation (Figure 6), in case the donor does not carry the genetic variant and has a normal complement phenotype.
Therefore, it is mandatory to perform a complete genetic screen in donors5,117.
If related donor and the recipient share any genetic factor of susceptibility for aHUS or if mutations have not been identified in the recipient, living donor transplantation should not be attempted5,117,128. In addition, living donor transplantation should only be considered if eculizumab is available5.
Terminal complement blockage in renal transplantation
Individual assessments of the recurrence risk based on complement investigations and recurrence in previous grafts may be used to guide the therapeutic strategy in renal transplant candidates (Table 3).
Currently, it is recommended that patients at high risk of aHUS recurrence should receive prophylaxis with eculizumab prior to surgery and thereafter (pre-emptive)5,6,16.
The efficacy and security of such a prophylactic regimen was studied in a series of treated French patients were absence of recurrence as well preservation or renal function are reported128, as well in case-reports, mainly in the pediatric setting89,129-132.
In patients having not received prophylaxis with eculizumab, blockade of terminal complement may still be used as a high efficacious rescue treatment for aHUS post-transplant recurrence (on-demand)128. Treatment for the recurrence of aHUS in patients receiving renal transplants should be performed similarly as for aHUS affecting native kidneys5,128.
Relevant for this discussion is the publication of a recent cost-effectiveness analysis using a decision analytical approach that was used to compare alternatives for aHUS patients with ESRD133, namely; 1) dialysis treatment, 2) kidney transplantation, 3) kidney transplantation with eculizumab therapy upon recurrence of aHUS, 4) kidney transplantation with eculizumab induction consisting of 12 months of prophylaxis and 5) kidney transplantation with lifelong eculizumab prophylaxis.
The authors concluded demonstrated that adding eculizumab resulted in a substantial gain in quality-adjusted life years (QALYS) compared to dialysis, but when compared to eculizumab administered upon recurrence, neither the drug induction nor lifelong eculizumab prophylaxis resulted in more QALYs, but did increase the overall costs. Therefore, the authors concluded that giving eculizumab upon recurrence of aHUS is more acceptable133.
Also, Duineveld et al134 describe their experience with kidney transplantation in patients with aHUS, without prophylactic eculizumab. They used a protocol with living kidney donors to limit ischemia-reperfusion injury, together with an immunosuppressive regimen comprising basiliximab, prednisone, high-dose mycophenolate mofetil, and tacrolimus dosed to target lower-than-usual blood concentrations. Sixteen of the 17 patients treated were at high risk for posttransplantation recurrence. During follow-up ranging from 7 to 68 months after transplantation, only 1 patient treated with their protocol had clinical signs of recurrence.
Eculizumab treatment in this patient induced hematologic remission but only a partial improvement in transplant function. Although these results open the discussion if posttransplantation eculizumab therapy should change from a prophylaxis strategy to a rescue approach, several limitations were pointed, namely the small cohort size and relatively short-term follow-up.
In spite of those analysis133 and data134 the working group recommends that until further data is presented, recommendation of national and international societies should be followed5,6,16 namely, prescribing eculizumab based on (genetic) risk stratification analysis.
Eculizumab is also effective in treating aHUS in patients with non CFH mutations5,128; therefore it is predicted that the response to eculizumab was independent of mutation status. Recurrence does occur in patients in the absence of an identified abnormality and therefore these patients have to be considered at medium risk of recurrence. Patients with no identifiable mutation have been shown to respond to eculizumab treatment128.
Another important question is the optimal duration of prophylactic eculizumab therapy after transplantation.
In patients with high-risk mutations or who have experienced a recurrence in a previous graft, anti-C5 therapy should not be discontinued117,128,135.
In patients with moderate risk, prophylactic treatment may be discontinued after a long recurrence-free period117, being patients closely monitored.
The working group recommends (Figures 4, 5, 6 and Table 3):
a) The genetic profile of the complement system is used to stratify the risk of aHUS recurrence after transplantation and to predict graft survival.
b) The decision to use anti-complement therapy during transplantation should be based on recurrence risk.
c) Patients with high or moderate risk of recurrence should be offered prophylactic eculizumab treatment.
d) Prophylactic eculizumab therapy should be recommended in patients with a high risk of posttransplantation aHUS recurrence and should be initiated prior to surgery at day 0 and include an additional dose at day 1.
e) Patients at low risk should be warned of the risk of recurrence and monitored closely.
f) There is a relative contraindication to living related donation but this can be considered in certain conditions, and only be considered if eculizumab is available. Kidney transplantation remains inadvisable if the donor shares a genetic susceptibilityfactor with the recipient or if no mutations have been identified in complement genes.
g) In the absence of evidence that CNI usage increases the risk of recurrent aHUS, these drugs should be included in immunosuppressive post-transplant protocols.
h) Tacrolimus is recommended due to the lower rate of post-transplant TMA reported.
i) The use of anti-IL2 receptor blocking antibody, anti-proliferative agent and steroids should be as local protocols.
j) mTOR inhibitors should be avoided in post-transplant in patients at risk of recurrent aHUS.
CONCLUSIONS AND FUTURE PERSPECTIVES
Twenty years have elapsed between the characterization of the first CFH mutations in aHUS patients and the widespread availability of a highly efficient therapy for this entity. During that period, translational research programs that brought together physicians and biomedical researchers, molecular biologists and immunologists in particular, have clarified the alternative complement pathway dysregulation that underlies aHUS.
And in the end, it was biological plausibility that enabled the introduction of a successful targeted therapy.
But target therapy is not equivalent to personalized medicine and, as such, a major a concern for clinicians is prescribing eculizumab for patients that may no longer benefit from terminal complement blockage. In order to settle this, an assay that reliably assesses disease activity is in great demand. Such a test has to be reproducible and easy to implement in routine immunology labs. A reported ex vivo endothelial complement deposition assay that requires cell culture facilities only available in research or academic institutions is, for example, an unrealistic option136. Such a test will help us in establishing treatment duration, since it became clear that in a significant fraction of patients, terminal complement blockage needs not to be prescribed for life. Finally, and considering what has happened with other recombinant molecules for medical use, eculizumab biosimilars, pegylated or long-acting formulations, as well monoclonal, peptides or small chemically synthetized compounds targeted to upstream complement mediators are expected to enter the market.
Since costs and patient convenience always impact physicians practice, the way those will change current guidelines is unpredictable (for details check aHUS pipeline 2017 at http://www.ahusallianceaction.org/ahus-therapeutic-drugs-research-development/).
It is now apparent that abnormalities of alternative complement pathway play a role in a larger fraction of renal diseases than what was initially suspected, somehow overshadowing the importance of the more traditional (and) classical pathway. They range from the inconspicuous role in renal disease progression mediated by acidosis, in which disruption of the internal thioester bond of C3 by ammonia promotes tubulointerstitial fibrosis137, to IgA nephropathy, where deletion of the CFHR1-3 locus was found to be protective138, or ANCA associated vasculitis for which C3 hypocomplementemia is associated with worst renal prognosis139 and C5a receptor antagonism is being considered as a therapeutic option140. Probably the most proxy is, of course, C3 glomerulopathy (C3G). An increasing number of reports depicting abnormalities in the complement genes, mostly involving large rearrangements within the CFH-CFHR locus (with and without anti-fH antibodies), is being matched by similar increasing number of reports detailing the off-label use of eculizumab in C3G141.
Atypical Hemolytic Uremic Syndrome, a rare kidney disease, allowed the nephrology community to clinically explore and manipulate the complexity of the complement system in the best interest of our patients. Let us hope that the insights gained with this journey can expand our interventional knowledge on more prevalent nephropathies.
References
1. Dragon-Durey MA, Frémeaux-Bacchi V. Atypical haemolytic uraemic syndrome and mutations in complement regulator genes. Springer Semin Immunopathol. 2005;27(3):359-74. [ Links ]
2. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-79. [ Links ]
3. Thompson RA, Winterborn MH. Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol. 1981;46(1):110-9. [ Links ]
4. I. AP. Soliris: summary of product characteristics 2012. [ Links ]
5. Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421-47. [ Links ]
6. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-51. [ Links ]
7. Kato H, Nangaku M, Hataya H, Sawai T, Ashida A, Fujimaru R, et al. Clinical guides for atypical hemolytic uremic syndrome in Japan. Clin Exp Nephrol. 2016;20(4):536-43. [ Links ]
8. NICE. Eculizumab for treating atypical haemolytic uraemic syndrome. 2015. [ Links ]
9. Sakari Jokiranta T, Viklicky O, Al Shorafa S, Coppo R, Gasteyger C, Macia M, et al. Differential diagnosis of thrombotic microangiopathy in nephrology. BMC Nephrol. 2017;18(1):324. [ Links ]
10. Symmers WS. Thrombotic microangiopathic haemolytic anaemia (thrombotic microangiopathy). Br Med J. 1952;2(4790):897-903. [ Links ]
11. Jennette JC, Heptinstall RH. Heptinstalls pathology of the kidney. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. [ Links ]
12. Román E, Mendizábal S, Jarque I, de la Rubia J, Sempere A, Morales E, et al. Secondary thrombotic microangiopathy and eculizumab: A reasonable therapeutic option. Nefrologia. 2017;37(5):478-91. [ Links ]
13. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. [ Links ]
14. Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 2008;112(1):11-8. [ Links ]
15. Scully M, Hunt BJ, Benjamin S, Liesner R, Rose P, Peyvandi F, et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol. 2012;158(3):323-35. [ Links ]
16. Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15-39. [ Links ]
17. George JN, Charania RS. Evaluation of patients with microangiopathic hemolytic anemia and thrombocytopenia. Semin Thromb Hemost. 2013;39(2):153-60. [ Links ]
18. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-86. [ Links ]
19. CDC – Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food – 10 states, 2009. Morb Mortal Wkly Rep. 2010;59(14):418-22. [ Links ]
20. Ardissino G, Salardi S, Colombo E, Testa S, Borsa-Ghiringhelli N, Paglialonga F, et al. Epidemiology of haemolytic uremic syndrome in children. Data from the North Italian HUS network. Eur J Pediatr. 2016;175(4):465-73. [ Links ]
21. Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med. 2011;365(19):1771-80. [ Links ]
22. Egan JA, Bandarenko N, Hay SN, Paradowski L, Goldberg R, Nickeleit V, et al. Differentiating thrombotic microangiopathies induced by severe hypertension from anemia and thrombocytopenia seen in thrombotic thrombocytopenia purpura. J Clin Apher. 2004;19(3):125-9. [ Links ]
23. Spinale JM, Ruebner RL, Kaplan BS, Copelovitch L. Update on Streptococcus pneumonia associated hemolytic uremic syndrome. Curr Opin Pediatr. 2013;25(2):203-8. [ Links ]
24. Banerjee R, Hersh AL, Newland J, Beekmann SE, Polgreen PM, Bender J, et al. Streptococcus pneumoniae-associated hemolytic uremic syndrome among children in North America. Pediatr Infect Dis J. 2011;30(9):736-9. [ Links ]
25. Copelovitch L, Kaplan BS. Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol. 2008;23(11):1951-6. [ Links ]
26. Allen JC, McCulloch T, Kolhe NV, Kolh NV. Adult hemolytic uremic syndrome associated with Streptococcus pneumoniae. Clin Nephrol. 2014;82(2):144-8. [ Links ]
27. Song D, Wu LH, Wang FM, Yang XW, Zhu D, Chen M, et al. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther. 2013;15(1):R12. [ Links ]
28. Bienaimé F, Legendre C, Terzi F, Canaud G. Antiphospholipid syndrome and kidney disease. Kidney Int. 2017;91(1):34-44. [ Links ]
29. Woodworth TG, Suliman YA, Li W, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-91. [ Links ]
30. George JN. Systemic malignancies as a cause of unexpected microangiopathic
hemolytic anemia and thrombocytopenia. Oncology (Williston Park). 2011;25(10): 908-14.
31. Sharma AP, Greenberg CR, Prasad AN, Prasad C. Hemolytic uremic syndrome (HUS) secondary to cobalamin C (cblC) disorder. Pediatr Nephrol. 2007;22(12):2097-103. [ Links ]
32. Menni F, Testa S, Guez S, Chiarelli G, Alberti L, Esposito S. Neonatal atypical hemolytic uremic syndrome due to methylmalonic aciduria and homocystinuria. Pediatr Nephrol. 2012;27(8):1401-5. [ Links ]
33. Cornec-Le Gall E, Delmas Y, De Parscau L, Doucet L, Ogier H, Benoist JF, et al. Adult-onset eculizumab-resistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am J Kidney Dis. 2014;63(1):119-23. [ Links ]
34. Navarro D, Azevedo A, Sequeira S, Ferreira AC, Carvalho F, Fidalgo T, et al. Atypical adult-onset methylmalonic acidemia and homocystinuria presenting as hemolytic uremic syndrome. CEN Case Rep. 2018;7(1):73-6. [ Links ]
35. Nogueira C, Aiello C, Cerone R, Martins E, Caruso U, Moroni I, et al. Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab. 2008;93(4):475-80. [ Links ]
36. Dashe JS, Ramin SM, Cunningham FG. The long-term consequences of thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome) in pregnancy. Obstet Gynecol. 1998;91(5 Pt 1):662-8. [ Links ]
37. Fakhouri F, Vercel C, Frémeaux-Bacchi V. Obstetric nephrology: AKI and thrombotic microangiopathies in pregnancy. Clin J Am Soc Nephrol. 2012;7(12):2100-6. [ Links ]
38. George JN, Nester CM, McIntosh JJ. Syndromes of thrombotic microangiopathy associated with pregnancy. Hematology Am Soc Hematol Educ Program. 2015;2015:644-8. [ Links ]
39. Fakhouri F, Roumenina L, Provot F, Sallée M, Caillard S, Couzi L, et al. Pregnancyassociated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21(5):859-67. [ Links ]
40. Bruel A, Kavanagh D, Noris M, Delmas Y, Wong EKS, Bresin E, et al. Hemolytic Uremic Syndrome in Pregnancy and Postpartum. Clin J Am Soc Nephrol. 2017;12(8):1237-47. [ Links ]
41. Huerta A, Arjona E, Portoles J, Lopez-Sanchez P, Rabasco C, Espinosa M, et al. A retrospective study of pregnancy-associated atypical hemolytic uremic syndrome. Kidney Int. 2018;93(2):450-9. [ Links ]
42. Fakhouri F. Pregnancy-related thrombotic microangiopathies: Clues from complement biology. Transfus Apher Sci. 2016;54(2):199-202. [ Links ]
43. Kelly RJ, Höchsmann B, Szer J, Kulasekararaj A, de Guibert S, Röth A, et al. Eculizumab in Pregnant Patients with Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2015;373(11):1032-9. [ Links ]
44. Servais A, Devillard N, Frémeaux-Bacchi V, Hummel A, Salomon L, Contin-Bordes C, et al. Atypical haemolytic uraemic syndrome and pregnancy: outcome with ongoing eculizumab. Nephrol Dial Transplant. 2016;31(12):2122-30. [ Links ]
45. Scully M, Thomas M, Underwood M, Watson H, Langley K, Camilleri RS, et al. Thrombotic thrombocytopenic purpura and pregnancy: presentation, management, and subsequent pregnancy outcomes. Blood. 2014;124(2):211-9. [ Links ]
46. Keir L, Coward RJ. Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy. Pediatr Nephrol. 2011;26(4):523-33. [ Links ]
47. Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, et al. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016;89(2):278-88. [ Links ]
48. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-62. [ Links ]
49. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-59. [ Links ]
50. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: State of the art. Mol Immunol. 2015;67(1):31-42. [ Links ]
51. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18(8):2392-400. [ Links ]
52. Mathern DR, Heeger PS. Molecules great and small: the complement system. Clin J Am Soc Nephrol. 2015;10(9):1636-50. [ Links ]
53. Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53(4):836-44. [ Links ]
54. Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet. 2001;68(2):485-90. [ Links ]
55. Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100(22):12966-71. [ Links ]
56. Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41(6):e84. [ Links ]
57. Kavanagh D, Kemp EJ, Mayland E, Winney RJ, Duffield JS, Warwick G, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(7):2150-5. [ Links ]
58. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(2):555-63. [ Links ]
59. Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21(12):2180-7. [ Links ]
60. De Vriese AS, Sethi S, Van Praet J, Nath KA, Fervenza FC. Kidney Disease Caused by Dysregulation of the Complement Alternative Pathway: An Etiologic Approach. J Am Soc Nephrol. 2015;26(12):2917-29. [ Links ]
61. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25(1):55-64. [ Links ]
62. Conway EM. Thrombomodulin and its role in inflammation. Semin Immunopathol. 2012;34(1):107-25. [ Links ]
63. Almeida C, Sousa JM, Rocha R, Cerqueira L, Fanning S, Azevedo NF, et al. Detection of Escherichia coli O157 by peptide nucleic acid fluorescence in situ hybridization (PNAFISH) and comparison to a standard culture method. Appl Environ Microbiol. 2013;79(20):6293-300. [ Links ]
64. Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood. 2014;123(24):3733-8. [ Links ]
65. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(19):1847-8. [ Links ]
66. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17): 1676-87. [ Links ]
67. Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12(24):3385-95. [ Links ]
68. Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, López-Trascasa M, Sánchez-Corral P, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14(5):703-12. [ Links ]
69. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24(3):475-86. [ Links ]
70. Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115(2):379-87. [ Links ]
71. Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3(3):e41. [ Links ]
72. Phillips EH, Westwood JP, Brocklebank V, Wong EK, Tellez JO, Marchbank KJ, et al. The role of ADAMTS-13 activity and complement mutational analysis in differentiating acute thrombotic microangiopathies. J Thromb Haemost. 2016;14(1):175-85. [ Links ]
73. Simeoni I, Stephens JC, Hu F, Deevi SV, Megy K, Bariana TK, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016;127(23):2791-803. [ Links ]
74. Bu F, Borsa NG, Jones MB, Takanami E, Nishimura C, Hauer JJ, et al. High-Throughput Genetic Testing for Thrombotic Microangiopathies and C3 Glomerulopathies. J Am Soc Nephrol. 2016;27(4):1245-53. [ Links ]
75. Martin-Fernandez L, Marco P, Corrales I, Pérez R, Ramírez L, López S, et al. The Unravelling of the Genetic Architecture of Plasminogen Deficiency and its Relation to Thrombotic Disease. Sci Rep. 2016;6:39255. [ Links ]
76. Matthijs G, Souche E, Alders M, Corveleyn A, Eck S, Feenstra I, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. 2016;24(1):2-5. [ Links ]
77. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. [ Links ]
78. Fidalgo T, Martinho P, Pinto CS, et al. Combined study of ADAMTS13 and complement genes in the diagnosis of thrombotic microangiopathies using next‐generation sequencing. Res Pract Thromb Haemost; 2017. p. 69‐80. [ Links ]
79. Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1(1):88-99. [ Links ]
80. Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transplant. 2008;12(6):619-29. [ Links ]
81. Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3(10):e431. [ Links ]
82. Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013;13(3):663-75. [ Links ]
83. Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C. New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol. 2011;7(1):23-35. [ Links ]
84. Chan MR, Thomas CP, Torrealba JR, Djamali A, Fernandez LA, Nishimura CJ, et al. Recurrent atypical hemolytic uremic syndrome associated with factor I mutation in a living related renal transplant recipient. Am J Kidney Dis. 2009;53(2):321-6. [ Links ]
85. Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int. 2010;77(4):339-49. [ Links ]
86. Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. 2010;10(7):1517-23. [ Links ]
87. Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med.2009;361(4):345-57. [ Links ]
88. Sinibaldi S, Guzzo I, Piras R, Bresin E, Emma F, Dello Strologo L. Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant. 2013;17(8):E177-81. [ Links ]
89. Matar D, Naqvi F, Racusen LC, Carter-Monroe N, Montgomery RA, Alachkar N. Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation. 2014;98(11):1205-12. [ Links ]
90. Bambauer R, Latza R, Schiel R. Therapeutic apheresis in the treatment of hemolytic uremic syndrome in view of pathophysiological aspects. Ther Apher Dial. 2011;15(1):10-9. [ Links ]
91. Bambauer R, Latza R, Burgard D, Schiel R. Therapeutic Apheresis in Immunologic Renal and Neurological Diseases. Ther Apher Dial. 2017;21(1):6-21. [ Links ]
92. Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36(6):673-81. [ Links ]
93. Viatel, P. Syndrome hémolytique et urémique de ladulte traité avec succès par plasmaphérèses massives. J Urol Nephrol; 1979. [ Links ]
94. Ariceta G, Besbas N, Johnson S, Karpman D, Landau D, Licht C, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24(4):687-96. [ Links ]
95. Loirat C, Noris M, Fremeaux-Bacchi V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2008;23(11):1957-72. [ Links ]
96. McLeod BC, Sniecinski I, Ciavarella D, Owen H, Price TH, Randels MJ, et al. Frequency of immediate adverse effects associated with therapeutic apheresis. Transfusion. 1999;39(3):282-8. [ Links ]
97. Michon B, Moghrabi A, Winikoff R, Barrette S, Bernstein ML, Champagne J, et al. Complications of apheresis in children. Transfusion. 2007;47(10):1837-42. [ Links ]
98. Boyer O, Balzamo E, Charbit M, Biebuyck-Gougé N, Salomon R, Dragon-Durey MA, et al. Pulse cyclophosphamide therapy and clinical remission in atypical hemolytic uremic syndrome with anti-complement factor H autoantibodies. Am J Kidney Dis. 2010;55(5):923-7. [ Links ]
99. Sana G, Dragon-Durey MA, Charbit M, Bouchireb K, Rousset-Rouvière C, Bérard E, et al. Long-term remission of atypical HUS with anti-factor H antibodies after cyclophosphamide pulses. Pediatr Nephrol. 2014;29(1):75-83. [ Links ]
100. Khandelwal P, Gupta A, Sinha A, Saini S, Hari P, Dragon Durey MA, et al. Effect of plasma exchange and immunosuppressive medications on antibody titers and outcome in anti-complement factor H antibody-associated hemolytic uremic syndrome. Pediatr Nephrol. 2015;30(3):451-7. [ Links ]
101. Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85(5):1151-60. [ Links ]
102. Lionet A, Provôt F, Glowacki F, Frémeaux-Bacchi V, Hazzan M. A case of adult atypical haemolytic uraemic syndrome related to anti-factor H autoantibodies successfully treated by plasma exchange, corticosteroids and rituximab. NDT Plus. 2009;2(6):458-60. [ Links ]
103. Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14(12):857-77. [ Links ]
104. Thurman JM, Le Quintrec M. Targeting the complement cascade: novel treatments coming down the pike. Kidney Int. 2016;90(4):746-52. [ Links ]
105. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360(5):544-6. [ Links ]
106. Nürnberger J, Philipp T, Witzke O, Opazo Saez A, Vester U, Baba HA, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360(5):542-4. [ Links ]
107. Legendre CM, Licht C, Loirat C. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369(14):1379-80. [ Links ]
108. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-81. [ Links ]
109. Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, et al. Terminal Complement Inhibitor Eculizumab in Adult Patients With Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial. Am J Kidney Dis. 2016;68(1):84-93. [ Links ]
110. Brocklebank V, Kavanagh D. Complement C5-inhibiting therapy for the thrombotic microangiopathies: accumulating evidence, but not a panacea. Clin Kidney J. 2017;10(5):600-24. [ Links ]
111. DGS. Recomendações sobre vacinas extra programa nacional de vacinação. 2018. [ Links ]
112. DGS- Norma nº 016/2016 de 16/12/2016 aa. Programa Nacional de Vacinação 2017. 2017. [ Links ]
113. McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High Risk for Invasive Meningococcal Disease Among Patients Receiving Eculizumab (Soliris) Despite Receipt of Meningococcal Vaccine. MMWR Morb Mortal Wkly Rep. 2017;66(27):734-7. [ Links ]
114. Parikh SR, Lucidarme J, Bingham C, Warwicker P, Goodship T, Borrow R, et al. Meningococcal B Vaccine Failure With a Penicillin-Resistant Strain in a Young Adult on Long-Term Eculizumab. Pediatrics. 2017;140(3). [ Links ]
115. BULLETIN T, CH50. [Available from: https://www.wako-chemicals.de/files/download/pdf/ch50_technical_bulletin_110216_0211d1_gb.pdf [ Links ]
116. Macia M, de Alvaro Moreno F, Dutt T, Fehrman I, Hadaya K, Gasteyger C, et al. Current evidence on the discontinuation of eculizumab in patients with atypical haemolytic uraemic syndrome. Clin Kidney J. 2017;10(3):310-9. [ Links ]
117. Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V, aHUS/C3G FSGf. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8(11):643-57. [ Links ]
118. Povey H, Vundru R, Junglee N, Jibani M. Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol. 2014;82(5):326-31. [ Links ]
119. Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87(5):1061-73. [ Links ]
120. Noris M, Ruggenenti P, Remuzzi G. Kidney transplantation in patients with atypical hemolytic uremic syndrome: a therapeutic dilemma (or not)? Am J Kidney Dis. 2017;70(6):754-7. [ Links ]
121. Zarifian A, Meleg-Smith S, Odonovan R, Tesi RJ, Batuman V. Cyclosporine-associated thrombotic microangiopathy in renal allografts. Kidney Int. 1999;55(6):2457-66. [ Links ]
122. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4(2):481-508. [ Links ]
123. Reynolds JC, Agodoa LY, Yuan CM, Abbott KC. Thrombotic microangiopathy after renal transplantation in the United States. Am J Kidney Dis. 2003;42(5):1058-68. [ Links ]
124. Robson M, Côte I, Abbs I, Koffman G, Goldsmith D. Thrombotic micro-angiopathy with sirolimus-based immunosuppression: potentiation of calcineurin-inhibitor-induced endothelial damage? Am J Transplant. 2003;3(3):324-7. [ Links ]
125. Artz MA, Steenbergen EJ, Hoitsma AJ, Monnens LA, Wetzels JF. Renal transplantation in patients with hemolytic uremic syndrome: high rate of recurrence and increased incidence of acute rejections. Transplantation. 2003;76(5):821-6. [ Links ]
126. Quan A, Sullivan EK, Alexander SR. Recurrence of hemolytic uremic syndrome after renal transplantation in children: a report of the North American Pediatric Renal Transplant Cooperative Study. Transplantation. 2001;72(4):742-5. [ Links ]
127. Donne RL, Abbs I, Barany P, Elinder CG, Little M, Conlon P, et al. Recurrence of hemolytic uremic syndrome after live related renal transplantation associated with subsequent de novo disease in the donor. Am J Kidney Dis. 2002;40(6):E22. [ Links ]
128. Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12(12):3337-54. [ Links ]
129. Nester C, Stewart Z, Myers D, Jetton J, Nair R, Reed A, et al. Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2011;6(6):1488-94. [ Links ]
130. Zimmerhackl LB, Hofer J, Cortina G, Mark W, Würzner R, Jungraithmayr TC, et al. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N Engl J Med. 2010;362(18):1746-8. [ Links ]
131. Weitz M, Amon O, Bassler D, Koenigsrainer A, Nadalin S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr Nephrol. 2011;26(8):1325-9. [ Links ]
132. Krid S, Roumenina LT, Beury D, Charbit M, Boyer O, Frémeaux-Bacchi V, et al. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant. 2012;12(7):1938-44. [ Links ]
133. van den Brand JA, Verhave JC, Adang EM, Wetzels JF. Cost-effectiveness of eculizumab treatment after kidney transplantation in patients with atypical haemolytic uraemic syndrome. Nephrol Dial Transplant. 2017;32(suppl_1):i115-i22. [ Links ]
134. Duineveld C, Verhave JC, Berger SP, van de Kar NCAJ, Wetzels JFM. Living donor kidney transplantation in atypical hemolytic uremic syndrome: a case series. Am J Kidney Dis. 2017;70(6):770-7. [ Links ]
135. Noris M, Remuzzi G. Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens. 2013;22(6):704-12. [ Links ]
136. Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124(11):1715-26. [ Links ]
137. Raphael KL. Metabolic acidosis and subclinical metabolic acidosis in CKD. J Am Soc Nephrol. 2018;29(2):376-82. [ Links ]
138. Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321-7. [ Links ]
139. Deshayes S, Aouba A, Khoy K, Mariotte D, Lobbedez T, Martin Silva N. Hypocomplementemia is associated with worse renal survival in ANCA-positive granulomatosis with polyangiitis and microscopic polyangiitis. PLoS One. 2018;13(4):e0195680. [ Links ]
140. Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017;28(9):2756-67. [ Links ]
141. Barbour TD, Ruseva MM, Pickering MC. Update on C3 glomerulopathy. Nephrol Dial Transplant. 2016;31(5):717-25. [ Links ]
Ana Azevedo, MD
Hospital de Vila Franca de Xira
Rua Calouste Gulbenkian 1, 2600-009 Vila Franca de Xira
E-mail: azevedo.gomes.ana@gmail.com
Disclosure of potential conflicts of interest:
Ana Azevedo has received fees from Alexion for participating in expert meetings and teaching courses and is executive editor of the Portuguese Journal of Nephrology and Hypertension; Joaquim Calado has received fees from Alexion for participating in expert meetings and teaching courses and is CME editor of the Portuguese Journal of Nephrology and Hypertension; Josefina Santos has received fees from Alexion for participating in expert meetings and teaching courses and is deputy editor of Portuguese Journal of Nephrology and Hypertension;
Received for publication: Sep 19, 2018
Accepted in revised form: Sep 26, 2018
ANNEXES
Annex 1:
Annex 2:
Annex 3:
Annex 4:


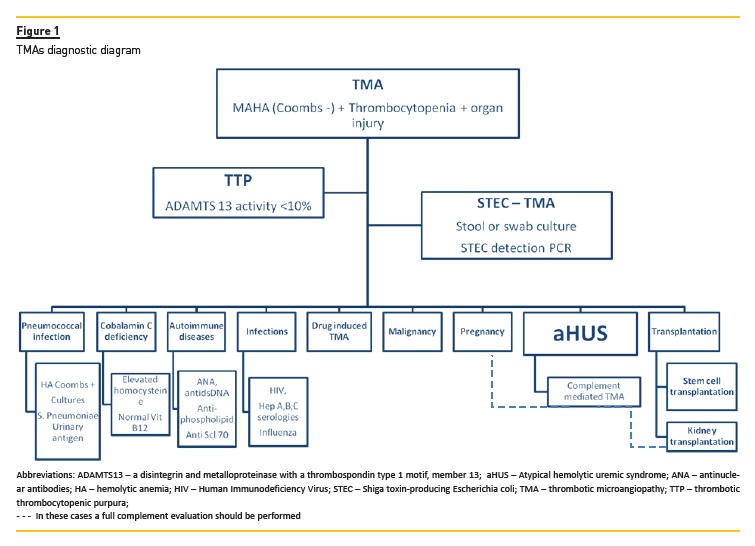{kind=link}
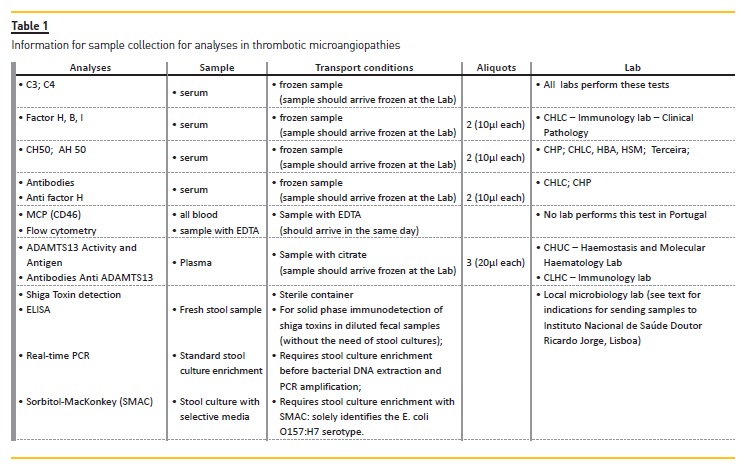{kind=link}
{kind=link}
{kind=link}
{kind=link}
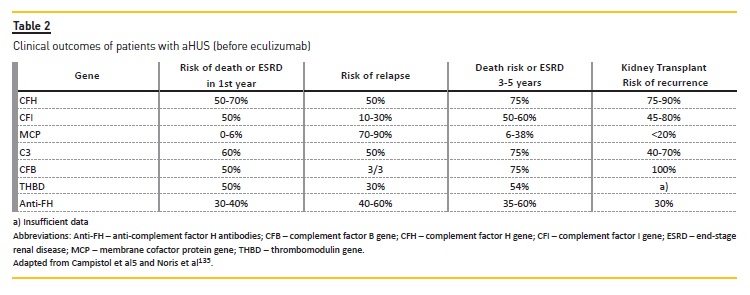{kind=link}
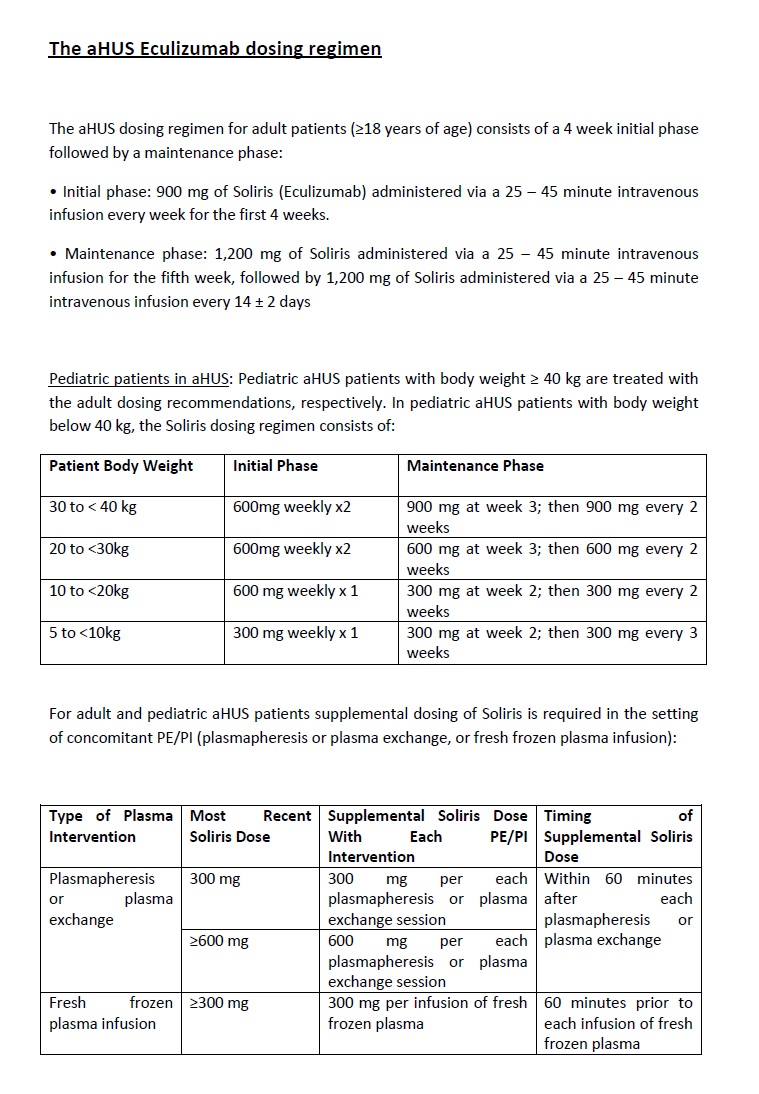{kind=link}
{kind=link}
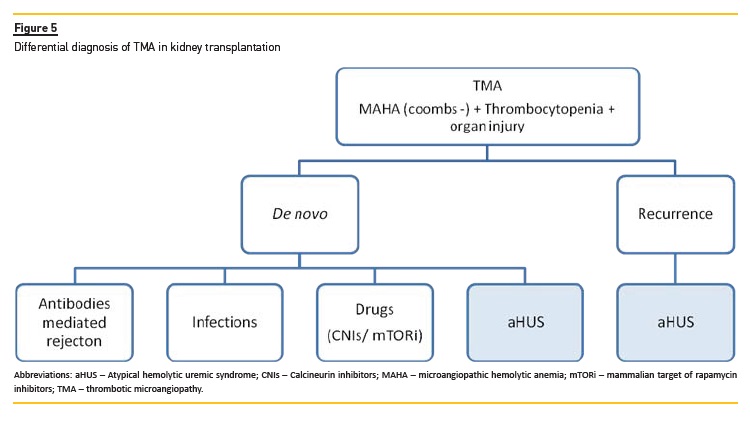{kind=link}
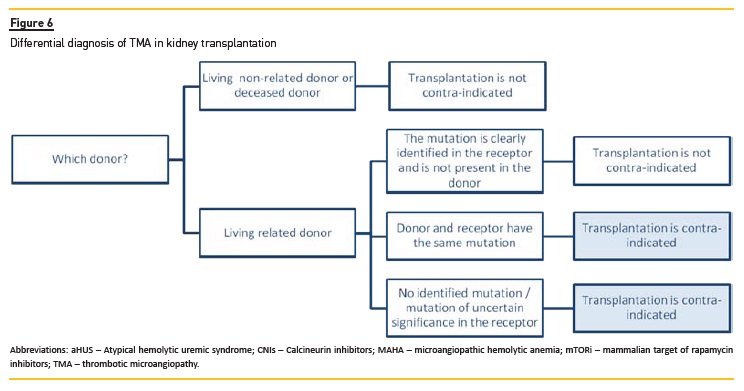{kind=link}