










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.33 no.1 Lisboa mar. 2019
https://doi.org/10.32932/pjnh.2019.04.010
CASE REPORT
Recurrence of oxalate nephropathy after renal transplantation in a patient with polyarteritis nodosa: a case report
Luís Mendonça1, Manuela Bustorf1, Roberto Silva2, Joana Sobrinho Simões3, Susana Sampaio1,4
1 Department of Nephrology, São João Hospital Center, EPE, Porto, Portugal.
2 Department of Pathology, São João Hospital Center, EPE, Porto, Portugal.
3 Department of Clinical Pathology, São João Hospital Center, EPE, Porto, Portugal.
4 Institute of Biomedical Engineering (INEB) – Institute of Investigation and Innovation in Health (i3S), Nephrology and Infectious Diseases Research and Development Group, University of Porto.
ABSTRACT
Hyperoxaluria is an important complication of short bowel syndrome (SBS) and a non‑negligible cause of chronic kidney disease (CKD). We describe the case of a 56‑year‑old woman with polyarteritis nodosa (PAN) and SBS who was admitted for acute kidney injury (AKI) 2 months after a successful deceased donor kidney transplantation. The diagnostic investigation was consistent with the diagnosis of oxalate nephropathy (OxN) and a set of lifestyle and therapeutic measures was implemented aiming at the reduction of intestinal oxalates absorption. Despite the short‑term benefit of this strategy, a progressive and inexorable deterioration of renal function was observed. In this report, we discuss the limited therapeutic options, the ominous prognosis and the risk of recurrence of this rare entity after renal transplantation. Additionally, we reflect on the value of patient compliance in the successful management of the disease and review potential new strategies that may improve the clinical course of this entity in the future.
Keywords: Hyperoxaluria; short bowel syndrome; kidney transplantation; polyarteritis nodosa.
INTRODUCTION
Hyperoxaluria is characterized by an increased urinary excretion of oxalate1. It is primarily caused by an impaired hepatic metabolism (i.e. primary hyperoxaluria) or an excessive absorption of oxalate from the intestine (i.e. secondary hyperoxaluria)1. The disease spectrum extends from recurrent kidney stones, nephrocalcinosis and urinary tract infections to chronic kidney disease (CKD) and end stage renal disease (ESRD)1.
We describe the case of a patient with polyarteritis nodosa (PAN) and short bowel syndrome (SBS) who presented a recurrence of oxalate nephropathy (OxN) a few weeks after kidney transplantation. Additionally, we discuss the management and the prognosis of this rare entity.
CASE PRESENTATION
Clinical History
A 56‑year‑old woman with ESRD on hemodialysis was admitted for deceased donor kidney transplantation. Her past medical was remarkable for PAN which had been diagnosed 10 years before after she presented acute monoarthritis of the knee, cutaneous vasculitis and ischemic nephropathy secondary to chronic thrombosis of the left renal artery and focal infarction of the right kidney. This clinical picture was preceded by an acute mesenteric thrombosis with evidence of vasculitis seen in the histopathologic exam 6 months earlier. In this setting, the patient was submitted to an extensive jejunum and ileum resection which caused chronic diarrhea and SBS. The cutaneous and rheumatologic manifestations were successfully treated with corticosteroids without recurrence. However, her renal disease progressed rapidly after the intestinal loss due to frequent episodes of renal colic and obstructive acute kidney injury (AKI). She started hemodialysis at the age of 49. Her course on dialysis was complicated by multiple access problems, mainly the primary failure of various arteriovenous fistulas caused by thrombosis. In this context, a thrombophilia screening revealed a double heterozygotic mutation on the methylenetetrahydrofolate reductase (MTHFR) gene of uncertain pathogenicity. Nonetheless, the patient was kept on warfarin since that time.
She was also an active smoker with a past history of a bladder carcinoma in situ successfully treated with local surgery and intravesical mitomycin. Her family history was unremarkable for renal disease.
The patient received a three‑antigen mismatch, 44‑year‑old deceased donor kidney transplant whose past medical history was unremarkable for relevant diseases. The panel reactive assay by complement‑dependent cytotoxicity was 0%. The transplant surgery had no complications and a cutaneous ureterostomy with a mono‑J catheter was decided on because she had an atrophic bladder. She received basiliximab for induction immunosuppression and her maintenance immunosuppression included prednisolone tapered to 5 mg per day six months after transplant; tacrolimus with dose adjusted to obtain trough levels between 4 and 7 ng/ml in the first two months and then between 2 and 5 ng/ml, and everolimus with dose adjusted to obtain trough levels between 3 and 8 ng/ml. The introduction of everolimus was driven by the potential antioncogenic effects of mTOR inhibitors and the lower risk of gastrointestinal symptoms when compared to mycophenolate mofetil.
The immediate post‑transplant period was complicated by a urinary tract infection and mechanical obstruction of the mono‑J, which were successfully treated with piperacillin‑tazobactam and replacement of the catheter. The patient did not undergo hemodialysis after transplantation.
Thereafter, her renal function progressively improved, and she was discharged with a serum creatinine of 0.98 mg/dl. Considering that the patient had SBS with chronic diarrhea, a set of measures aiming at the prevention of oxalate‑induced nephropathy was instituted, including high fluid intake (2‑3L/day), a diet poor in oxalates, calcium supplements (calcium carbonate 2g during meals) and urinary alkalinization (potassium citrate 6g/day). However, the lack of compliance with this plan was noted during the follow‑up visits, particularly regarding the diet. Two months after discharge, she was admitted with fever and oliguria.
Initial clinical approach
The physical exam was significant for tenderness over the allograft on abdominal palpation and 2+ pitting bilateral lower limb edema below the knee. The cardiac and pulmonary auscultation were unremarkable. The patient was febrile (tympanic temperature 38.5°C); blood pressure, heart rate and respiratory rate were normal (105/71 mmHg; 98 beats per minute and 12 breaths per minute, respectively).
The blood test revealed anemia (hemoglobin of 9.5 g/dL), slightly increased white blood cell counts (12.500/uL), increased C‑reactive protein (120 mg/dL), increased creatinine (4.6 mg/dl) increased urea (123 mg/dL), normal ionogram, normal corrected calcium (4.3 mEq/L), normal phosphorus (3.3 mg/dL), slightly decreased magnesium (1.4 mEq/L) and normal uric acid (6 mg/dL). The urinary sediment was remarkable for leukocyturia. Her renal ultrasound showed moderate hydronephrosis (renal pelvis with 27 mm) and the allograft vascular prefusion was preserved on doppler evaluation. The mono‑J catheter was obstructed with calculi and urine output recovered promptly after its replacement. Empiric antibiotic therapy with vancomycin was started according to a previous urine culture positive for Enterococcus faecium.
Her condition improved during the first week with sustained apyrexia, edema improvement, decreasing of serum inflammatory markers and maintenance of a good urine output. The urine culture confirmed the positivity for Enterococcus faecium and vancomycin was continued for 2 weeks. Blood cultures were negative. However, as her serum creatinine persisted above 3 mg/dL, she underwent a renal biopsy to exclude additional causes of allograft dysfunction.
Renal allograft biopsy
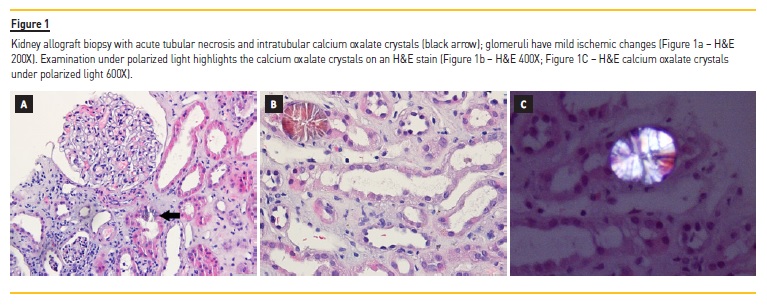
The histological examination of the allograft kidney biopsy (Figure 1) displayed scattered intratubular calcium oxalate crystals consistent with OxN. There was also acute tubular necrosis, moderate arterial fibroelastosis and interstitial fibrosis with tubular atrophy (20%). Glomeruli had mild ischemic changes and there were no signs of acute cellular or antibody mediated rejection. Immunofluorescence for C4d in peritubular capillaries was negative.
Additional Investigations
Donor specific antibodies were absent. The BK virus screening was negative in both blood and urine. A 24‑hour urine showed a high excretion of oxalates (128 mg/24h, normal range < 45 mg). A stool real‑time polymerase chain reaction searching for Oxalobacter formigenes was negative (Oxalyl‑CoA decarboxylase gene, Genesig®).
Clinical follow‑up
A plan towards oxalate reduction in urine was intensified, particularly focusing on the patient’s lifestyle. Her renal function recovered progressively with a serum creatinine of 2.2 mg/dl at discharge. The patient was regularly followed at the outpatient clinic and adhered to all the conservative measures for oxalate control. One year after the transplantation, urinary sediment showed rare oxalate crystals; 24‑hour urine had 65 mg of oxalates and no further hospital admissions for AKI were documented. However, a slow increase in serum creatinine was evident. In this setting, an additional workup excluded hydronephrosis as well as compromised allograft perfusion or significant proteinuria, and screening for BK virus or donor specific antibodies remained negative. We assumed progression of the allograft dysfunction secondary to oxalate nephropathy. Alternative therapeutic strategies were considered, particularly the use of total parenteral nutrition; however, given the difficulty in obtaining a good vascular access for central venous catheter insertion and the patient’s reluctance, this approach was deferred. In addition, we considered enrolling the patient in a clinical trial evaluating an oxalate decarboxylase analog.
However, her posttransplant status made her ineligible. Two years after transplantation, she was admitted for recurrence of bladder carcinoma. At that time, serum creatinine was 3.5 mg/dL. The bladder resection surgery was complicated with acute kidney injury and continuous renal replacement therapy was needed. The patient died of septic shock after one week.
DISCUSSION
Oxalate is the ionic form of oxalic acid and is derived from various animal and plant sources2. It is eliminated mainly through the kidneys and normal daily excretion in healthy individuals ranges from 10‑40 mg per 24 h2. Increased urinary quantities may be seen in patients with SBS and this problem became more evident with the advent of modern bariatric surgery3. In the small intestine, malabsorption and steatorrhea cause intraluminal calcium to bind preferentially with free fatty acids, leaving oxalates free to be absorbed2. Other contributing factors include colonic mucosal alterations, metabolic acidosis, a concentrated urine from chronic diarrhea, reduced urinary concentration of magnesium and citrate, and changes in intestinal microbiota2. CKD and ESRD constitute important complications of hyperoxaluria4. In our patient, the ischemic loss of the small intestine and secondary oxalate nephrolithiasis certainly accelerated the progression of her previous renal disease associated with PAN. Additionally, the absence of Oxalobacter formigenes might also be deleterious in this setting, given that this bacterium degrades oxalates from diet through the oxalate decarboxylase.
In ESRD associated with SBS, renal transplantation is challenged by several factors, including the risk of recurrence and the malabsorption of immunosuppressive drugs5. Only a few cases have been reported with recognized variable success5‑9.
The implementation of a preventable strategy is the rule6, which is limited and significantly dependent on patient habits, as we have shown6. The use of drugs such as cholestyramine, although not contraindicated, requires an additional care in intake timing, given potential interference with the absorption of immunosuppressive drugs10,11. In this setting, new therapies are being studied, such as the modulation of oxalate degradation or gut microbiome1. Oxalate decarboxylase has shown promising results in animal models and is currently being tested in humans12.
However, as shown, transplanted patients have not been included in these studies13. Roodnat et al. have recently published a small study with 10 living‑donor kidney recipients who received intense hemodialysis to lower plasma oxalic acid levels before and immediately after renal transplantation and favorable short term outcomes were observed in most of them9. Our patient did not undergo hemodialysis after transplantation; however, her urine output was good. In addition, the unpredictability of deceased donor kidney transplantation did not allow a long‑controlled period of enteric oxalate reduction before transplantation.
Ceulemans et al. reported the success of a combined kidney and intestinal transplantation in two patients with enteric hyperoxaluria14.
Although this surgery would theoretically correct the primary disturbance of these patients, a recent case of unexpected relapse associated with a molecular defect in the anion exchange transporter SLC26A6 has shown that the intestinal hyperabsorption of oxalates is probably still unrecognized15.
In addition to the cohort mentioned above, a few more cases of OxN recurrence have been published and most of them were observed in patients with inflammatory bowel disease9. The prognosis was variable and included long‑term excellent allograft function, histological recurrence with normal renal function and early graft loss after transplantation6,9. Our case is the first to be reported in a patient with SBS secondary to a vascular PAN complication. In our opinion, this case raises some relevant questions regarding the clinical management of OxN in transplanted patients. First, the clinical value of urinary oxalates quantification and its correlation with renal outcomes.
Our patient had high urinary oxalates a few days after transplantation, which decreased about 50% over one year. Although this biochemical improvement may represent a true reduction in intestinal oxalates, this interpretation should be adjusted for renal function. It has been demonstrated that the plasma oxalate level starts increasing at a GFR of about 30 ml/min and oxalate retention becomes significant when the GFR decreases below about 20 ml/min16. The quantification of plasma oxalate concentration could probably constitute a more accurate indicator, although its use is not available in many centers, as is the case in ours. So, the value of the quantification of urinary oxalates can be misleading and, in fact, the renal function of our patient maintained a downward trend despite the apparent metabolic improvement. Although a kidney biopsy was not repeated, it is plausible to assume a diagnosis of oxalate nephropathy progression, considering the setting presented and the exclusion of alternative diagnosis such as vascular thrombosis or ureteral obstruction. In addition, the plasma levels of the immunosuppressive drugs were stable within the therapeutic range, and no DSA were identified. Some previous cases showed this weak correlation between urinary oxalates and renal outcomes6,7. Our case also emphasizes the importance of the “second‑hit” concept9.
It has been discussed that an acute intercurrence, such as an infection or dehydration, may disturb oxalic acid homeostasis and result in temporary partial or complete loss of renal function in these particularly vulnerable patients. As described, our patient experienced her first AKI and recognized recurrence of OxN when she had an acute pyelonephritis. We only observed a partial restoration of renal function, but sometimes an additional recovery is seen later. On this topic, Cuvelier et al. reported a surprising recovery of graft function in a patient with chronic pancreatitis after 11 months on hemodialysis8.
Finally, this case also shows that the successful management of the disease is significantly conditioned by patient´s compliance with therapy and daily habits, mainly regarding nutrition. This topic should be thoroughly discussed in a pre‑transplant setting because the lack of a strict compliance will certainly aggravate the prognosis of these patients when transplanted.
In conclusion, we report a rare case of OxN after kidney transplantation in a patient with enteric hyper oxaluria secondary to a vascular complication of PAN. Currently, the disease is essentially dependent upon the control of oxalate absorption through conservative measures. New therapies are important to improve the prognosis of these patients.
References
1. Bhasin B, Urekli HM, Atta MG. Primary and secondary hyperoxaluria: Understanding the enigma. World J Nephrol. 2015;4(2):235‑44. [ Links ]
2. Marengo SR, Romani AM. Oxalate in renal stone disease: the terminal metabolite that just won’t go away. Nat Clin Pract Nephrol. 2008;4(7):368‑77.
3. Troxell ML, Houghton DC, Hawkey M, Batiuk TD, Bennett WM. Enteric oxalate nephropathy in the renal allograft: an underrecognized complication of bariatric surgery. Am J Transplant. 2013;13(2):501‑9. [ Links ]
4. Hassan I, Juncos LA, Milliner DS, Sarmiento JM, Sarr MG. Chronic renal failure secondary to oxalate nephropathy: a preventable complication after jejunoileal bypass. Mayo Clin Proc. 2001;76(7):758‑60. [ Links ]
5. Roberts RA, Sketris IS, MacDonald AS, Belitsky P. Renal transplantation in secondary oxalosis. Transplantation. 1988;45(5):985‑6. [ Links ]
6. Nazzal L, Puri S, Goldfarb DS. Enteric hyperoxaluria: an important cause of end‑stage kidney disease. Nephrol Dial Transplant. 2016;31(3):375‑82. [ Links ]
7. Rifkin SI. Transplantation for renal failure secondary to enteric hyperoxaluria: a case report. J Med Case Rep. 2007;1:31. [ Links ]
8. Cuvelier C, Goffin E, Cosyns JP, Wauthier M, de Strihou C. Enteric hyperoxaluria: a hidden cause of early renal graft failure in two successive transplants: spontaneous late graft recovery. Am J Kidney Dis. 2002;40(1):E3. [ Links ]
9. Roodnat JI, de Mik‑van Egmond AME, Visser WJ, Berger SP, van der Meijden WAG, Knauf F, et al. A successful approach to kidney transplantation in patients with enteric (secondary) hyperoxaluria. Transplant Direct. 2017;3(12):e331. [ Links ]
10. Mignat C. Clinically significant drug interactions with new immunosuppressive agents. Drug Saf. 1997;16(4):267‑78. [ Links ]
11. Jensen RA, Lal SM, Diaz‑Arias A, James‑Kracke M, Van Stone JC, Ross G, Jr. Does cholestyramine interfere with cyclosporine absorption? A prospective study in renal transplant patients. ASAIO J. 1995;41(3):M704‑6.
12. Langman CB, Grujic D, Pease RM, Easter L, Nezzer J, Margolin A, et al. A double‑blind, placebo controlled, randomized phase 1 cross‑over study with ALLN‑177, an orally administered oxalate degrading enzyme. Am J Nephrol. 2016;44(2):150‑8. [ Links ]
13. Evaluate ALLN‑177 in Patients With Enteric Hyperoxaluria [Available from: https://ClinicalTrials.gov/show/NCT03456830. [ Links ]
14. Ceulemans LJ, Nijs Y, Nuytens F, De Hertogh G, Claes K, Bammens B, et al. Combined kidney and intestinal transplantation in patients with enteric hyperoxaluria secondary to short bowel syndrome. Am J Transplant. 2013;13(7):1910‑4. [ Links ]
15. Ekser B, Mangus RS, Kubal CA, Fridell JA, Powelson JA, Nagaraju S, et al. Recurrence of Hyperoxaluria and Kidney Disease after Combined Intestine‑Kidney
Transplantation for Enteric Hyperoxaluria. Am J Nephrol. 2016;44(2):85‑91.
16. Jonassen JA, Kohjimoto Y, Scheid CR, Schmidt M. Oxalate toxicity in renal cells. Urol Res. 2005;33(5):329‑39. [ Links ]
Luís Carlos Ferreira Mendonça
Hospital S. João, Alameda Professor Hernâni Monteiro, 4200 Porto, Portugal
E‑mail: luiscfmendonca@gmail.com
Disclosure of potential conflicts of interest: none declared.
Received for publication: Dec 22, 2018
Accepted in revised form: Feb 28, 2019


{kind=link}