










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.33 no.2 Lisboa jun. 2019
https://doi.org/10.32932/pjnh.2019.07.026
CASE REPORT
Nephronophthisis, a pediatric challenge
Ana M. Garcia, Sónia M. Gomes, Telma Francisco, Ana P. Serrão
Paediatric Nephrology Unit, Department of Paediatrics Hospital de Dona Estefânia, Centro Hospital Universitário de Lisboa Central, EPE
ABSTRACT
Nephronophthisis is an autosomal recessive cystic kidney disease characterized by reduced concentrating ability of the kidney, chronic tubulointerstitial nephritis, cystic renal disease and progression to end-stage renal disease. In 10-20% of cases it is associated with a variety of other ciliopathy phenotypes.
A 10-year-old caucasian female patient presented to our hospital with a history of uncontrolled vomiting and signs of moderate dehydration.
Past medical history was significant for reduced visual acuity and moderate intellectual disability with a minor cerebellar dysplasia. She also complained of polyuria, polydipsia and enuresis for several years. Laboratory screening revealed a low urine specific gravity and renal insufficiency that were maintained after intravenous rehydration.
Based on the clinical picture, a ciliopathy was considered, particularly nephronophthisis with neurological manifestation and confirmed by genetic molecular analysis.
In conclusion, this case report reinforces the diagnostic challenge of nephronophthisis because of its unspecific presentation and significant overlap with other ciliopathy phenotypes. Therefore, in the presence of a urinary concentration defect and renal impairment, with or without other extra-renal manifestations, nephronophthisis should be considered in the differential diagnosis to allow a prompt diagnosis and therapeutic intervention.
Keywords: chronic kidney disease, ciliopathy, nephronophthisis, urinary concentration defect.
INTRODUCTION
Nephronophthisis (NPH) was first described by Smith and Graham in 1945 and was termed “medullary cystic kidney disease”1; the current term “nephronophthisis” derives from Greek and means “disintegration of nephron”, also an aspect of the histopathology.2
It is an autosomal recessive cystic kidney disease and is the most frequent genetic cause of end‑stage renal disease (ESRD) in children and young adults.3 The estimated incidence varies worldwide, from 1:50 000 liveborns in Finland and Canada to 1:1 000 000 in the Unites States of America, although this data may be underreported.4 NPH is a genetically heterogeneous disorder with more than 20 genes identified.5 Almost all encode proteins expressed in centrosomes and primary cilia; therefore NHP can be considered a ciliopathy disease.3,4,6,7
Renal phenotype is characterized by reduced concentrating ability of the kidney, chronic tubulointerstitial nephritis, cystic renal disease and progression to ESRD.7
Patients present initially with polyuria, polydipsia and enuresis.8 The onset of the disease may be easily missed, as there is typically no severe hypertension, minimal or no proteinuria and a bland urine sediment. Furthermore, in 10‑20% of cases it is associated with a variety of other ciliopathy phenotypes.2,9,10 Therefore, a high index of suspicion is necessary for diagnosing NPH.
CASE REPORT
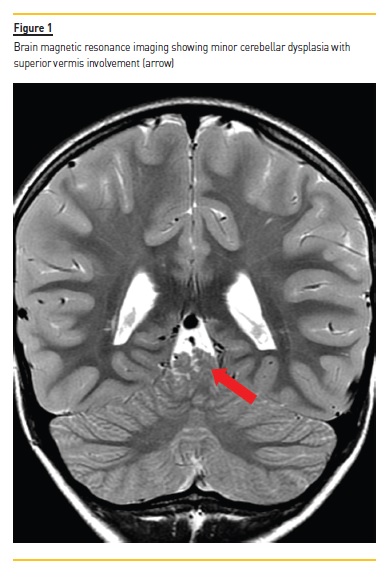
A 10‑year‑old caucasian female patient presented to our hospital with a history of uncontrolled vomiting. Past medical history was significant for reduced visual acuity (hyperopia and astigmatism); moderate intellectual disability, disrupt motor coordination and hypotonia of unknown etiology, for which she was under neurological care. She also had a minor cerebellar dysplasia with superior vermis involvement on brain magnetic resonance imaging (Figure 1). At six‑years old, she was referred to nephrology consultation for suspected hypertension, which was not confirmed, and at that time renal function was normal. Her family history was unremarkable concerning renal pathology, but both her parents had minor mental disability. There was no consanguinity. At physical examination, signs of moderate dehydration were present. Blood pressure was 97/53 mmHg (<90 percentile), cardiac frequency was 84 beats per minute, body weight was 24.2 Kg (3–15 percentile) and height was 138 cm (50 percentile).
Laboratory data were as follows (table I): hemoglobin 10.1 g/dL, hematocrit 27.9 %; blood urea nitrogen 142 mg/dL, creatinine 2.1 mg/dL, glomerular filtration rate 26.55mL/min/1,73m2; and blood gas pH 7.34, HCO3‑18.2, BE ‑7.7. Urinalysis showed a specific gravity of 1005 with no other alteration.
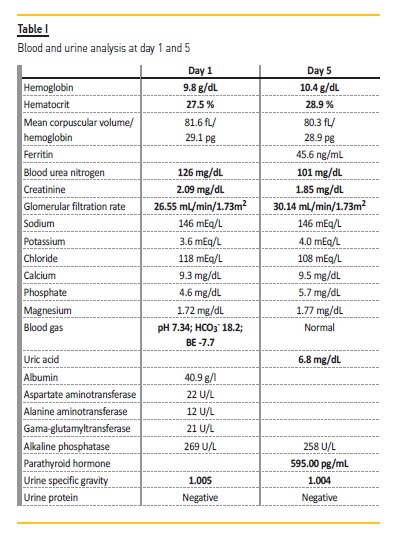
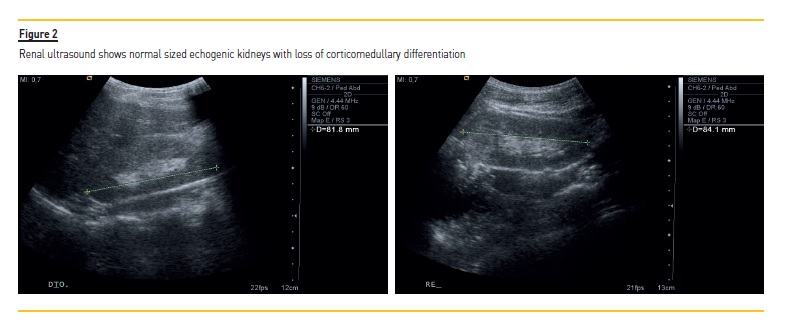
Acute kidney injury was suspected and hospital admission was prompt. After intravenous rehydration, renal function stabilized at high levels (blood urea nitrogen 101 mg/dL, creatinine 1.85 mg/dL, glomerular filtration rate of 30.14 mL/min/1.73m2) and urinary concentration defect persisted (urine specific gravity of 1004) (Table I). On detailed history, she complained of polyuria, polydipsia and enuresis for several years. Renal ultrasound revealed normal sized echogenic kidneys with loss of corticomedullary differentiation (Figure 2). Abdominal ultrasound revealed no abnormalities. Ophthalmologic examination was unremarkable besides refractory errors. Cardiologic evaluation was normal.
We suspected a diagnosis of ciliopathy particularly NPH with neurological manifestation. This hypothesis was confirmed by microarray‑based comparative genomic hybridization (aCGH), which revealed a biallelic 2q13 deletion of 482 kbp involving 14 genes including NPHP1 gene. The other genes included in the mutation are RGPD6, RGPD5, LIMS3L, LIMS3, LIMS3‑LOC440895, LOC440895, LOC100288570, LOC440894, MIR4267, MALL, MIR4436B2, MIR4436B1 and LINC00116.
The array used has a density of 550,000 oligonucleotides and 200,436 SNPs, in a total of 750,436 markers distributed throughout the genome, with a mean resolution of 8‑20 Kbp.
The genetic study was performed more than one year before the diagnosis of chronic kidney disease, and was ordered due to the neurological alterations, but the result was only available a few weeks after the admission of the patient on our ward. Renal biopsy was not performed.
She started conservative therapy for chronic kidney disease (cholecalciferol, calcitriol and iron) and her parents were referred for genetic counselling. The mutation was later identified in both parents, in heterozygosity.
DISCUSSION
NPH is an autosomal recessive inheritable kidney disease caused by mutations in several genes. Nearly all these genes code for components of the cilium‑centrosome complex; therefore NPH can be considered a renal manifestation of a ciliopathy disease.3,4,7,11 However, the numerous pathways involved in the pathogenesis of the disease are not fully understood. Therefore, further genetic investigation is crucial to understand the underlying molecular mechanisms and consequently allow a personalized therapeutic approach.5
NPH has historically been classified based on the age of onset of ESRD as infantile, juvenile, adolescent and late onset, which correspond to a median age of 112, 1313, 19 years14 and adulthood15, respectively.
Juvenile form is the most frequent and is characterized by clinical presentation during the first decade of life, as in our case study. Clinical manifestations result from loss of distal tubular function and posterior impairment of glomerular filtration.8,13 Therefore, patients initially present with mild symptoms, including polyuria, polydipsia and enuresis, and later in the course of the disease, anemia and growth impairment occur. In contrast to other renal diseases, patients usually do not develop arterial hypertension until the renal function is severely impaired.16 In our case, low urine specific gravity in a dehydrated patient suggests an underlying urinary concentration defect. Furthermore, renal function stabilization at high levels of serum urea and creatinine after rehydration suggested chronic kidney disease (CKD) and clinical manifestations of polyuria, polydipsia and enuresis suggested a tubular defect. According to glomerular filtration rate, CKD can be categorized as G3b.17
In juvenile NPH, renal ultrasound scan often displays normal or slightly reduced kidney size, increased echogenicity and loss of corticomedullary differentiation, as in our case report. If cysts are present, they are typically small, arise from the corticomedullary border region of the kidney and develop in later stages of the disease.5
In 10–20% of cases, NPH is associated with extrarenal manifestations in organs such as the retina, central nervous system or liver.2,9,10
The coexistence of specific multisystem features allows the identification of known syndromes. Two of the most frequently reported in literature are Senior‑Loken Syndrome (SLS) and Joubert Syndrome (JS). The first one describes the association of NPH with retinal involvement, namely retinitis pigmentosa, tapetoretinal degeneration or retinal aplasia.18,19 Usually, ophthalmic alterations are present in all SLS by the age of 10. The second one is a developmental disorder characterized by cerebellar vermis hypoplasia, which represents the radiological imaging hallmark of JS known as “molar tooth sign”.20
In our case, renal features associated with ocular and neurologic manifestations suggested a possible ciliopathy. However, absence of retinal changes and typical brain image excluded the previous recognizable syndromes. Normal liver function testing and abdominal ultrasound also excluded liver involvement. Therefore, we could only classify it as NHP with neurological disorder.
To date, the diagnosis of NPH is suggested by clinical features, renal ultrasound findings and family history and confirmed by molecular genetic approach.5 The most common genetic cause are mutations of the NPHP1 gene, which accounts for around 20% of cases, namely large homozygous deletion affecting the whole gene.4,13,21 Each of the remaining NPH genes probably account for 1% or less of all cases, and around two‑thirds remain without genetic diagnosis.4 There is little genotype‑phenotype correlation; therefore mutation in one gene may cause a wide spectrum of clinical manifestations.22 Consequently, NPHP1 gene mutation may be screened using standard PCR assays, but given the large number of other NPH genes involved, multiplex PCR, targeted exon capture or whole‑exome sequencing should be considered.23‑25
In our case, the diagnosis of NPH was made by performing aCGH, which revealed a biallelic 2q13 deletion of 482 kbp involving 14 genes and encompassing NPHP1 gene. Usually, this is not the first‑line approach when suspecting of NPH, and nowadays the diagnosis is more frequently established by performing Next Generation Sequencing (NGS) panels. However, as stated before in the description of the case, the CGH was initially ordered due to the neurological disability, without the knowledge of the chronic kidney disease, and later, when the patient was evaluated by our nephrology department and the diagnosis was clinically suspected, we decided to wait for the pending results, which confirmed our hypothesis. Further, an earlier paper by Haghighi26 reports a family in which the diagnosis of NPH was made using CGH.
The deletion found in our patient has a size of 482 kbp and involves 14 genes. Of these, only the deletion of NPHP1 is clinically relevant. Full gene deletions are recurrent in the general population because NPHP1 locus has some unique features that make this region very prone to non‑allelic homologous recombination. As shown by Saunier and colleagues21, NPHP1 deletion is 290 kb in size and NPHP1 is flanked by two large inverted repeats of ∼330 kb. These flanking low copy repeats are elements with highly similar sequence identities.27
A second sequence of 45 kb is located adjacent to the proximal 330‑kb repeat, which is directly repeated 250 kb away within the distal 330‑kb repeat deleting the sequence tag site (STS) 804H10R present in the proximal copy (an STS is a relatively short and easily PCR‑amplified sequence that occurs only once in the chromosome that is being studied and therefore is used in mapping the human genome). The deletion breakpoints are located within the 45‑kb repeat, suggesting an unequal recombination between the two homologous copies of this smaller repeat. Moreover, Saunier and colleagues also demonstrated in their study a nonpathologic rearrangement involving the two 330‑kb inverted repeats found in some patients and, in the homozygous state, in a few control individuals. This could be explained by interchromosomal mispairing of the 330‑kb inverted repeat, followed by double recombination or by a prior intrachromosomal mispairing of these repeats, leading to an inversion of the NPHP1 region, followed by an interchromosomal unequal crossover event.21 The deletion found in the majority of patients, as well as complex rearrangement explained previously demonstrates the high level of rearrangements occurring in this region. NPH is the most prevalent genetic for ESRD in children7, and most frequently (20‑25%) NPH is caused by deletions in the NPHP1 gene.28
Although it is considered a classic paediatric Mendelian kidney disease, Snoek et al27 described a prevalence of 0.5% of homozygous NPHP1 full gene deletions in adult‑onset ESRD. Considering that other mutation types were not analysed, this prevalence is likely underdiagnosed and probably is higher than previously reported.
Renal biopsy should be performed when genetic testing is negative or is not available. The hallmark histologic triad consists of a) tubular basement membrane disintegration and thickening; b) interstitial cell infiltration with fibrosis; c) tubular atrophy and cyst formation at the corticomedullary junction.16 In our case, molecular identification of the most common mutation superseded renal biopsy.
Despite intensive research, a curative or targeted therapy for NPH is not yet available. Treatment of NPH remains supportive and includes management of complications arising from progressive renal failure and preparation for future renal replacement therapy. Recurrence of NPH after renal transplant has never been reported.5
With regard to our patient´s prognosis, given the diagnosis of juvenile NPH, it will be expected to evolve to ESRD by early adolescence.13
In conclusion, this case report highlights the diagnostic challenge of NPH because of its unspecific presentation and significant overlap with other ciliopathy phenotypes. Therefore, in the presence of polydipsia, polyuria, anemia and renal impairment, NPH should be considered, to allow a prompt diagnosis and early therapeutic intervention.
References
1. Smith CH, Graham JB. Congenital medullary cysts of the kidneys with severe refractory anemia. Am J Dis Child 1945;69:369–77. [ Links ]
2. Wolf MTF, Hildebrandt F. Nephronophthisis. Pediatr Nephrol 2011;26:181‑94. [ Links ]
3. Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet 2005;6:928–40. [ Links ]
4. Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol 2009;20:23–35. [ Links ]
5. Srivastava S, Molinari E, Raman S, Sayer JA. Many genes – one disease? genetics of nephronophthisis (NPHP) and NPHP‑associated disorders. Front Pediatr 2018;5:1–15. [ Links ]
6. Simms RJ, Hynes AM, Eley L, Sayer JA. Nephronophthisis: a genetically diverse ciliopathy. Int J Nephrol 2011;2011:1–10. [ Links ]
7. Hildebrandt F, Zhou W. Nephronophthisis‑associated ciliopathies. J Am Soc Nephrol 2007;18:1855–71. [ Links ]
8. Krishnan R, Eley L, Sayer JA. Urinary concentration defects and mechanisms underlying nephronophthisis. Kidney Blood Press Res 2008;31:152–62. [ Links ]
9. Wolf MTF. Nephronophthisis and related syndromes. Curr Opin Pediatr 2015;27:201–11. [ Links ]
10. Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol 2009;24:2333–44. [ Links ]
11. Simms RJ, Eley L, Sayer JA. Nephronophthisis. Eur J Hum Genet 2009;17:406–16. [ Links ]
12. Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo‑interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol 1989;3:50–5. [ Links ]
13. Hildebrandt F, Strahm B, Nothwang HG, et al. Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. Kidney Int 1997;51:261–9. [ Links ]
14. Olbrich H, Fliegauf M, Hoefele J, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto‑retinal degeneration and hepatic fibrosis. Nat Genet 2003;34:455–9. [ Links ]
15. Hoefele J, Nayir A, Chaki M, et al. Pseudodominant inheritance of nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr Nephrol 2011;26:967–71. [ Links ]
16. Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol 2017;9:1–28. [ Links ]
17. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 2013;3:1–150. [ Links ]
18. Løken AC, Hanssen O, Halvorsen S, Jolster NJ. Hereditary Renal Dysplasia and Blindness. Acta Paediatr 1961;50:177–84. [ Links ]
19. Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophy. Am J Ophthalmol 1961;52:625–33. [ Links ]
20. Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet 2007;15:511–21. [ Links ]
21. Saunier S, Calado J, Benessy F, et al. Characterization of the NPHP1 locus: Mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet 2000;66:778–89. [ Links ]
22. König J, Kranz B, König S, et al. Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol 2017;12:1974–83. [ Links ]
23. Otto EA, Helou J, Allen SJ, et al. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat 2008;29:418–26. [ Links ]
24. Halbritter J, Diaz K, Chaki M, et al. High‑throughput mutation analysis in patients with a nephronophthisis‑associated ciliopathy applying multiplexed barcoded array‑based PCR amplification and next‑generation sequencing. J Med Genet 2012;49:756–67. [ Links ]
25. Gee HY, Otto EA, Hurd TW, et al. Whole‑exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int 2014;85:880–7. [ Links ]
26. Haghighi A, Savaj S, Haghighi‑Kakhki H, Benoit V, Grissart B, Dahan K. Identification of an NPHP1 delection causing adult form of nephronophthisis. Ir J Med Sci 2016;185:589–95. [ Links ]
27. Snoek R, van Setten J, Keating BJ et al. NPHP1 (Nephrocystin‑1) Gene Delections Cause Adult‑Onset ESRD. J Am Soc Nephrol 2018;29(6):1172–9. [ Links ]
28. Halbritter J, Porath JD, Diaz KA, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis‑related ciliopathy. Hum Genet 2013;132: 865–84. [ Links ]
Ana Margarida Marques Garcia
Paediatric Nephrology Unit, Department of Paediatrics
Hospital de Dona Estefânia, Centro Hospital Universitário de Lisboa Central, EPE
Rua Jacinta Marto, 1169‑045
Lisboa
E‑mail: garcia_anamargarida@hotmail.com
Disclosure of potential conflicts of interest: none declared.
Received for publication: Dec 9
Accepted in revised form: Jun 4


{kind=link}