










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.33 no.4 Lisboa dez. 2019
https://doi.org/10.32932/pjnh.2020.01.050
CASE REPORT
IgA Nephropathy with Thrombotic microangiopathy: Is this secondary thrombotic microangiopathy or IgA nephropathy-triggered atypical Hemolytic Uremic Syndrome?
Hugo Diniz1,2, Maria Bandeira3, Ana Teresa Nunes1, Bruno Besteiro4, João Coimbra4, Filipa Gomes4, Susana Sampaio1,2
1 Nephrology Department, Centro Hospitalar e Universitário de São João, Oporto, Portugal
2 Nephrology & Infectious Diseases R&D, i3S – Instituto de Investigação e Inovação em Saúde da Universidade do Porto, Oporto, Portugal
3 Université Catholique de Louvain, Faculté de Médecine et Médecine Dentaire, Brussels, Belgium
4 Internal Medicine Department, Centro Hospitalar e Universitário de São João, Oporto, Portugal
ABSTRACT
IgA Nephropathy (IgAN) is the most prevalent biopsy-proven primary glomerular disease worldwide. Historically, thrombotic microangiopathy (TMA) was associated with IgAN in cases of severe hypertension or advanced chronic kidney disease, but recent data suggest that complement activation plays a crucial role in the development of TMA in IgAN. We report a case of Crescentic IgAN with complement-mediated TMA (C-TMA) in a 27-year old male patient with a pathological missense mutation in heterozygosity in the CFH gene and a rare variant in the C3 gene, treated with steroids, cyclophosphamide and plasmapheresis without recovery of kidney function. We also discuss other treatment possibilities and kidney transplant options. Additionally, we will review the latest advances that are enhancing our understanding of the association between IgAN and TMA.
Keywords: Complement; Genetics; Hemolytic Uremic Syndrome; IgA Nephropathy; Thrombotic Microangiopathies.
INTRODUCTION
IgA Nephropathy (IgAN) is a glomerular disease characterized by diffuse deposits of IgA in the mesangial areas of the glomerulus and is the most common biopsy-proven primary glomerular disease worldwide1.
Thrombotic microangiopathy (TMA) is a heterogeneous disorder characterized by microangiopathic hemolytic anemia, thrombocytopenia, and organ injury caused by arteriolar and capillary thrombosis with characteristic abnormalities in the endothelium and vessel wall2.
Historically, TMA has been associated with IgAN in cases of severe hypertension or advanced chronic kidney disease (CKD)3,4, but emerging data is challenging this hypothesis5-7. Still, the prevalence and the pathophysiology of this association are poorly understood.
We report a case of an acute presentation of IgAN that was associated with TMA at the time of diagnosis. Additionally, we will discuss the latest advances that are enhancing our understanding of the association between IgAN and TMA.
CASE REPORT
A 27-year old male patient, with no relevant past medical history, was admitted to the emergency department due to pallor and asthenia. The patient had been in his usual state of health until approximately three weeks before admission, when he developed abdominal pain, nausea, vomiting, and diarrhea, treated with non-steroidal anti-inflammatory drugs. These symptoms lasted for five days. He reported long-term intake of protein supplement consumption, as well as other supplements, to increase his muscle mass until six months previously.
He denied having a fever, rash or purpura, epistaxis, red-eye, dyspnea, cough, or lumbar pain. He had no familiar history of kidney disease. On clinical examination, the patient was pale and ill-appearing. His blood pressure was 170/90 mmHg; heart rate 76 bpm; oxygen saturation 95%, and he was eupneic and afebrile. Cardiovascular examination revealed a mild proto-systolic murmur at the apex. He had peripheral bilateral edema, and there was no evidence of rash or palpable purpura. Diuresis was maintained.
In the emergency department, his laboratory exams revealed severe anemia (Hb 5.7 g/dl), with increased reticulocyte count to 3.5% (0.5–2.5 %), discrete thrombocytopenia (139.000 x10^9/uL), and someschizocytes (1-2/high-power field). They also revealed severe azotemia (serum urea 233 mg/dl; serum creatinine 11.62 mg/dl), increased LDH (962 U/L), and normal c-reactive protein (3.9 mg/dl). Blood gases revealed a metabolic acidosis (pH 7.33 HCO3 15.3 mmol/L). The Coombs direct test was negative, clotting study unremarkable, and D-dimers were normal (3.47 g/ml). Urinalysis revealed proteinuria (1.5 g/L), leucocyturia, and erythrocyturia. Renal ultrasound showed normally dimensioned kidneys.
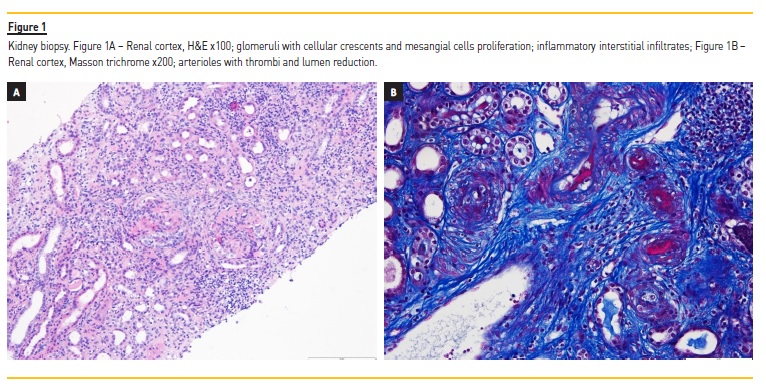
The patient was admitted to the ward to start hemodialysis, after receiving two units of packed red blood cells. A kidney biopsy was performed on the next day. The anatomopathological analysis revealed 25 glomeruli, 14 of which had cellular crescents and 7 were sclerosed.
There were signs of thrombotic microangiopathy, tubular necrosis, hematuria, and moderate interstitial lymphocytic infiltrate (Figure 1).
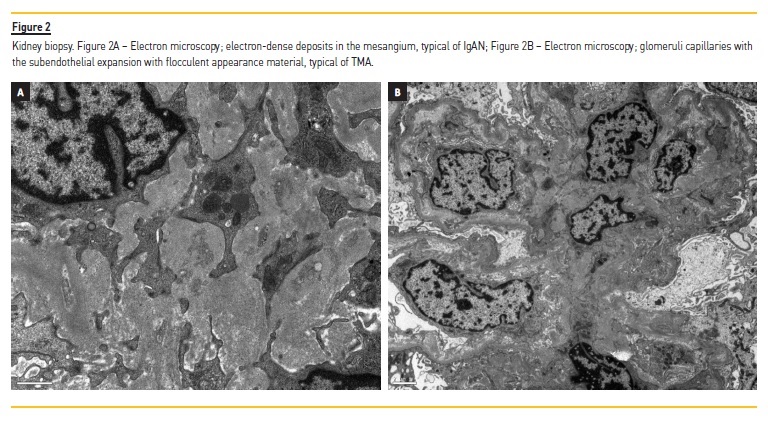
Immunofluorescence showed mesangial deposits of IgA (++). Further workup showed proteinuria 2.05 g/24 h, decreased haptoglobin (8 mg/dl) and increased serum IgA (341 mg/dl). The virologic and immunological study was negative.
A diagnosis of crescentic IgAN with TMA was assumed, and the patient started on steroids (3 pulses of intravenous methylprednisolone 500 mg and then prednisolone 1 mg/kg per os), and pulsed intravenous cyclophosphamide 1000 mg.
Due to the presence of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and TMA in the kidney biopsy, further work-up was undertaken, and plasmapheresis with fresh plasma was started. ADAMTS13 enzyme activity was normal (89%), and ADAMTS13 inhibitor and anti-factor H antibodies were negative. The ultrastructural study showed a diffuse expansion of the lamina rara interna, with flocculent appearance, revealing the dimension of the TMA involvement (Figure 2).
The molecular study of the complement genes revealed a missense pathological mutation in heterozygosity in the complement factor-H (CFH) gene (c.3611G>A, p.Gly1204Glu) for complement-mediated TMA (C-TMA) and a rare missense variant in heterozygosity in the C3 gene (c.463A>C, p.Lys155Gln), usually benign but associated with atypical Hemolytic Uremic Syndrome (aHUS) when co-inherited with pathogenic variants. After ten sessions of plasmapheresis, there was no evidence of microangiopathic hemolytic anemia, and the patient was discharged on regular hemodialysis through a central tunneled catheter. There was no improvement in kidney function, despite four intravenous pulses of cyclophosphamide and daily prednisone. After the results of the molecular study of the complement genes, it was decided to stop the cyclophosphamide pulses, and the steroids were quickly tapered. A final diagnosis of crescentic IgA Nephropathy with complement-mediated thrombotic microangiopathy was considered. The patient is in a continuous ambulatory peritoneal dialysis program and is being followed in the pre-transplant clinic.
DISCUSSION
Crescentic IgAN is an uncommon presentation of IgAN, representing approximately 5% of all IgAN cases and is associated with poor renal prognosis8. On the other hand, crescentic IgAN with C-TMA has rarely been reported in the literature9,10.
The CFH mutation identified in our patient is highly pathogenic, with a score of 5/5 in silico, and affects the short consensus repeat 20 (SCR20) of the carboxyterminal region of factor H (FH), causing a substitution of a glycine residue that reduces the capacity of FH to bind to heparin11.
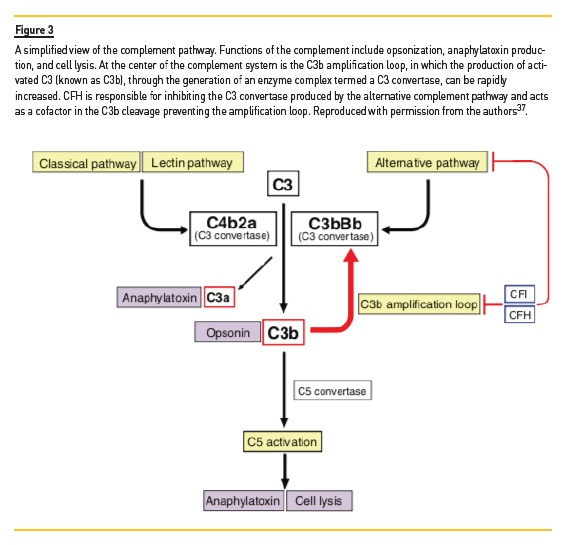
This region is critical for the attachment of FH to the surface of host cells and consequently for the regulation of the complement alternative pathway activation12. Upon an inflammatory insult, the defective cell recognition of factor H reduces its regulatory activities of the alternative pathway and the C3b amplification loop at the surface of endothelial cells, which results in cell damage and exposure of the subendothelial matrix, causing C-TMA (Figure 3)13. Patients with CFH mutations have the worst outcome of all the patients with C-TMA, usually progressing to end-stage renal disease (ESRD) or death within one year of presentation14. The C3 variant is usually benign but can have a cumulative effect on the patients phenotype when co-inherited with pathologic mutations15.
In the presented case, the rapidly progressive renal insufficiency was preceded by acute gastrointestinal symptoms three weeks before.
Both IgA vasculitis and aHUS can be associated with gastrointestinal manifestations, but acute gastroenteritis, in a patient with genetic susceptibility, could have been the trigger of both the crescentic transformation of IgAN and C-TMA.
Infection is a known precipitant of C-TMA16. Dysregulated intestinal mucosal activation due to infection also increases serum IgA and its deposition in the mesangium1,17,18. This acute change in IgA leads to direct or indirect activation of the complement system7,19, endothelial dysfunction, and kidney damage. This link could explain the overlap of aHUS and IgAN in the setting of infection reported in the literature10.
In the past, it was thought that IgAN was only associated with TMA in cases of severe hypertension or advanced CKD4. Karoui et al.5 reported for the first time that TMA is a common histopathological feature of IgAN (53% of patients) and that TMA can occur in normotensive patients, or with near-normal renal histology. They also found out that TMA is associated with worse renal prognosis, even after adjusting for the hypertension severity. Recent Chinese and Dutch retrospective studies have reported similar findings. In the Chinese cohort, histological microangiopathic lesions were present in 20.6 % of IgAN biopsies, and 23.2% of the patients with microangiopathic lesions were normotensive7. In the Dutch cohort, histological microangiopathic lesions were present in 23% of IgAN biopsies, and 25% of the patients with microangiopathic lesions were normotensive6.
In both studies, histological microangiopathic lesions were associated with worse renal outcomes6,7. This data suggest that histological microangiopathic lesions are frequent in IgAN when both acute and chronic lesions are considered.
In the Dutch study, the kidney fragments were also restained for complement proteins using immunohistochemistry, including C4d and C5b-96. Microangiopathy was associated with C4d and C5b-9 deposits, a higher number of chronic lesions, and hypertension. This data suggests that local complement activation is involved in the development of microangiopathy in patients with IgAN, and hypertension does not seem to be the primary cause of TMA in IgAN. Interestingly, a strong association between mutations in complement genes and hypertension-associated TMA was also reported in another study20. This could mean that hypertension-associated TMA in IgAN is mediated by complement dysregulation as well.
Complement activation is also involved in the pathophysiology of IgAN. Mesangial IgA, mainly polymeric IgA1, drives the activation of the mesangial cells, which is amplified by the ability of IgA to activate the complement system through the lectin and alternative pathways (Figure 3)19,21. Genome-wide association studies (GWAS) in IgAN also hint to the role of deletions of CFH receptor (CHFR) 1, CFHR3, and CFHR5 variants for IgAN susceptibility18,22. In another study, five IgAN patients with low serum factor H were found to carry CFH or complement factor I (CFI) pathogenic variants, which suggests a decreased factor H activity in IgAN23. Whether or not there is a relationship between complement alterations in IgAN and the development of TMA remains to be proven, but there are several anecdotal reports of this association24,25. Ongoing trials are exploring the therapeutic effects of blocking the complement pathway in IgAN, for example, with avacopan, a C5a receptor blocker, and may also increase our understanding of the pathophysiology of IgAN26.
The final diagnosis of crescentic IgAN with C-TMA still leaves the question: is this a secondary TMA associated with crescentic IgAN, or is this an IgAN-triggered aHUS? The association of crescentic IgAN with histological TMA is a consequence of the intense endothelial lesion associated with the glomerulonephritis pathophysiology, similarly to what happens with other glomerulopathies like Lupus Nephritis.
Despite occurring a local complement activation, this usually does not cause MAHA. Our patient developed a systemic involvement with MAHA, which makes us consider that it was the Crescentic IgAN that triggered the aHUS, as a consequence of a mutated FH.
The Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend the use of steroids and cyclophosphamide for the treatment of Crescentic IgAN27. Given the extent of the TMA lesions on the kidney biopsy and the mutations in the CFH and C3 genes, eculizumab, an anti-C5 antibody, could have been an effective therapy. In fact, there are case reports of crescentic IgAN28,29 and IgAN with C-TMA treated with eculizumab with different results30,31. Genetic testing is a slow procedure, and our institution only approves eculizumab use if there is proof of pathogenic mutations associated with C-TMA or aHUS. Genetic testing results were reported approximately two months after we started taking care of this patient. At the time, there were no signs of kidney recovery despite treatment with steroids and cyclophosphamide, and we considered that starting eculizumab late in the course of the disease would be unfruitful.
Our patients genetic panel will also influence his future kidney transplant opportunities and prognosis. Most transplant centers would advise against related living-donation for a patient with C-TMA attributed to a genetic mutation. Although genetic analysis can be performed in donors, some patients may have more than one genetic variation, and approximately one-third of patients with C-TMA have yet-to-beidentified mutations32. Besides, nephrectomy may trigger C-TMA in a genetically susceptible donor33. Due to the risk of C-TMA recurrence after kidney transplantation, prophylactic therapy with eculizumab is recommended in patients with CFH or CFI mutations34,35. Unfortunately, the optimal regimen and duration of eculizumab therapy after transplantation is not known.
The absence of Shiga-toxin testing in a patient with MAHA and gastrointestinal symptoms can be pointed out as a limitation of our diagnostic work-up. This test was not ordered on admission because the patient had no dysentery, and diarrhea had resolved two weeks before his hospital admission. On the other hand, gastrointestinal symptoms have been observed in about one-quarter of the patients with C-TMA36.
In summary, we report a case of Crescentic IgAN with complement- mediated TMA in a 27-year old male patient with a pathological missense mutation in the CFH gene and a rare variant in the C3 gene, treated with steroids, cyclophosphamide and plasmapheresis without recovery of kidney function. Laboratory signs of MAHA in a young patient should prompt complement genetic testing even when accompanied by another clinical entity that can be associated with TMA.
References
1. Wyatt RJ, Julian BA. IgA Nephropathy. N Engl J Med [Internet]. 2013 Jun 20 [cited 2019 Jul 28];368(25):2402–14. Available from: http://www.nejm.org/doi/10.1056/NEJMra1206793 [ Links ]
2. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med [Internet]. 2014 Aug 14 [cited 2019 Jul 28];371(7):654–66. Available from: http://www.nejm.org/doi/10.1056/NEJMra1312353 [ Links ]
3. A Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Cattran DC, Coppo R, Cook HT, Feehally J, Roberts ISD, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int [Internet]. 2009 Sep [cited 2019 Jul 28];76(5):534–45. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0085253815540118 [ Links ]
4. Chang A, Kowalewska J, Smith KD, Nicosia RF, Alpers CE. A clinicopathologic study of thrombotic microangiopathy in the setting of IgA nephropathy. Clin Nephrol [Internet]. 2006 Dec 1 [cited 2019Aug 4];66(12):397–404. Available from: http://www.dustri.com/article_response_page.html?artId=959&doi=10.5414/CNP66397&L=0 [ Links ]
5. El Karoui K, Hill GS, Karras A, Jacquot C, Moulonguet L, Kourilsky O, et al. A clinicopathologic study of thrombotic microangiopathy in IgA nephropathy. J Am Soc Nephrol [Internet]. 2012 Jan [cited2019 Jul 28];23(1):137–48. Available from: http://www.jasn.org/lookup/doi/10.1681/ASN.2010111130 [ Links ]
6. Chua JS, Zandbergen M, Wolterbeek R, Baelde HJ, van Es LA, de Fijter JW, et al. Complementmediated microangiopathy in IgA nephropathy and IgA vasculitis with nephritis. Mod Pathol [Internet]. 2019 Jul [cited 2019 Aug 4];32(8):1147–57. Available from: http://www.nature.com/articles/s41379-019-0259-z. [ Links ]
7. Cai Q, Shi S, Wang S, Ren Y, Hou W, Liu L, et al. Microangiopathic lesions in IgA nephropathy: a cohort study. Am J Kidney Dis [Internet]. 2019 May [cited 2019 Aug 4]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S0272638619306638 [ Links ]
8. Abuelo JG, Esparza AR, Matarese RA, Endreny RG, Carvalho JS, Allegra SR. Crescentic IgA nephropathy. Medicine (Baltimore) [Internet]. 1984 Nov [cited 2019 Aug 4];63(6):396. Available from: https://insights.ovid.com/crossref?an=00005792-198411000-00005 [ Links ]
9. Sürmeli-Döven S, Delibaş A, Gürses İ, Kayacan UR, Coşkun-Yılmaz B, Esen K, et al. Hemolytic uremic syndrome and iga nephropathy in a child: coincidence or not? Turk J Pediatr [Internet]. 2018 [cited 2019 Aug 5];60(1):81. Available from: http://www.turkishjournalpediatrics.org/doi.php?doi=10.24953/turkjped.2018.01.012 [ Links ]
10. Wang R, Zhang Y, Li S, Chen H, Zeng C, Chen H, et al. Hemolytic uremic syndrome complicated with IgA nephropathy: a case report and literature review. Clin Nephrol [Internet]. 2015 Jan 1 [cited 2019 Aug 5];83 (2015)(01):36–40. Available from: http://www.dustri.com/article_response_page.html?artId=11075&doi=10.5414/CN108000&L=0. [ Links ]
11. Saunders RE, Abarrategui-Garrido C, Frémeaux-Bacchi V, Goicoechea de Jorge E, Goodship THJ, López Trascasa M, et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat [Internet]. 2007 Mar [cited 2019 Aug 5];28(3):222–34. Available from: http://doi.wiley.com/10.1002/humu.20435 [ Links ]
12. Józsi M, Heinen S, Hartmann A, Ostrowicz CW, Hälbich S, Richter H, et al. Factor H and atypical hemolytic uremic syndrome: mutations in the c-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol [Internet]. 2006 Jan [cited 2019 Oct 14];17(1):170–7. Available from: http://www.jasn.org/lookup/doi/10.1681/ASN.2005080868 [ Links ]
13. Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology. 2000 Aug;49(1–2):149–57. [ Links ]
14. Sellier-Leclerc A-L, Fremeaux-Bacchi V, Dragon-Durey M-A, Macher M-A, Niaudet P, Guest G, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol [Internet]. 2007 Aug [cited 2019 Aug 6];18(8):2392–400. Available from: http://www.jasn.org/lookup/doi/10.1681/ASN.2006080811 [ Links ]
15. Geerlings MJ, Volokhina EB, de Jong EK, van de Kar N, Pauper M, Hoyng CB, et al. Genotypephenotype correlations of low-frequency variants in the complement system in renal disease and age-related macular degeneration. Clin Genet [Internet]. 2018 Oct [cited 2019 Aug 5];94(3–4):330–8. Available from: http://doi.wiley.com/10.1111/cge.13392 [ Links ]
16. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship THJ, et al. Atypical aHUS: state of the art. Mol Immunol [Internet]. 2015 Sep [cited 2019 Aug 5];67(1):31–42. Available from:https://linkinghub.elsevier.com/retrieve/pii/S0161589015003508 [ Links ]
17. Emancipator SN. Experimental models of IgA nephropathy. Am J Kidney Dis [Internet]. 1988 Nov [cited 2019 Aug 5];12(5):415–9. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0272638688800374 [ Links ]
18. Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet [Internet]. 2014 Nov [cited 2019 Aug 5];46(11):1187–96. Available from: http://www.nature.com/articles/ng.3118 [ Links ]
19. Faria B, Henriques C, Matos AC, Daha MR, Pestana M, Seelen M. Combined C4d and CD3 immunostaining predicts immunoglobulin (Ig)A nephropathy progression: biomarkers of IgA nephropathy progression. Clin Exp Immunol [Internet]. 2015 Feb [cited 2019 Aug 4];179(2):354–61. Available from: http://doi.wiley.com/10.1111/cei.12461 [ Links ]
20. Timmermans SAMEG, Abdul-Hamid MA, Vanderlocht J, Damoiseaux JGMC, Reutelingsperger CP, van Paassen P, et al. Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities. Kidney Int [Internet]. 2017 Jun [cited 2019 Aug 4];91(6):1420–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0085253816307426 [ Links ]
21. Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy Is associated with more severe renal disease. J Am Soc Nephrol [Internet]. 2006 Jun [cited 2019 Aug 4];17(6):1724–34. Available from: http://www.jasn.org/lookup/doi/10.1681/ASN.2005090923 [ Links ]
22. Noris M, Remuzzi G. Genetics of immune-mediated glomerular diseases: focus on complement. Semin Nephrol [Internet]. 2017 Sep [cited 2019 Aug 4];37(5):447–63. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0270929517300578 [ Links ]
23. Tortajada A, Gutiérrez E, Goicoechea de Jorge E, Anter J, Segarra A, Espinosa M, et al. Elevated factor H–related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int [Internet]. 2017 Oct [cited 2019 Aug 5];92(4):953–63. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0085253817302545 [ Links ]
24. Schmitt R, Krmar RT, Kristoffersson A, Söderberg M, Karpman D. IgA nephropathy associated with a novel N-terminal mutation in factor H. Eur J Pediatr [Internet]. 2011 Jan [cited 2019 Aug 5];170(1):107–10. Available from: http://link.springer.com/10.1007/s00431-010-1279-3 [ Links ]
25. Jia M, Zhu L, Zhai Y, Chen P, Xu B, Guo W, et al. Variation in complement factor H affects complement activation in immunoglobulin A vasculitis with nephritis. Nephrology [Internet]. 2019 May 5 [cited 2019 Aug 4]; Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/nep.13580 [ Links ]
26. Selvaskandan H, Cheung CK, Muto M, Barratt J. New strategies and perspectives on managing IgA nephropathy. Clin Exp Nephrol [Internet]. 2019 May [cited 2019 Aug 6];23(5):577–88. Available from:http://link.springer.com/10.1007/s10157-019-01700-1 [ Links ]
27. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int Suppl [Internet]. 2012 Jun [cited 2019 Aug 5];2(2):142. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2157171615310480 [ Links ]
28. Ring T, Pedersen BB, Salkus G, Goodship THJ. Use of eculizumab in crescentic IgA nephropathy: proof of principle and conundrum? Clin Kidney J [Internet]. 2015 Oct [cited 2019 Aug 5];8(5):489–91. Available from: https://academic.oup.com/ckj/article-lookup/doi/10.1093/ckj/sfv076 [ Links ]
29. Rosenblad T, Rebetz J, Johansson M, Békássy Z, Sartz L, Karpman D. Eculizumab treatment for rescue of renal function in IgA nephropathy. Pediatr Nephrol [Internet]. 2014 Nov [cited 2019 Aug 5];29(11):2225–8. Available from: http://link.springer.com/10.1007/s00467-014-2863-y [ Links ]
30. Nakamura H, Anayama M, Makino M, Makino Y, Tamura K, Nagasawa M. Atypical hemolytic uremic syndrome associated with complement Factor H mutation and IgA nephropathy: a case report successfully treated with eculizumab. Nephron [Internet]. 2018 [cited 2019 Aug 5];138(4):324–7. Available from: https://www.karger.com/Article/FullText/485194 [ Links ]
31. Matsumura D, Tanaka A, Nakamura T, Sato E, Node K. Coexistence of atypical hemolytic uremic syndrome and crescentic IgA nephropathy treated with eculizumab: a case report. Clin Nephrol – Case Stud [Internet]. 2016 Dec 12 [cited 2019 Aug 5]; Available from: https://www.dustri.com/index.php?id=8&artId=14977&doi=10.5414/CNCS108889 [ Links ]
32. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009 Oct 22;361(17):1676–87. [ Links ]
33. Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transplant. 2008 Sep;12(6):619–29. [ Links ]
34. Levi C, Frémeaux-Bacchi V, Zuber J, Rabant M, Devriese M, Snanoudj R, et al. Midterm outcomes of 12 renal transplant recipients treated with eculizumab to prevent atypical hemolytic syndrome recurrence: transplantation [Internet]. 2017 Dec [cited 2019 Aug 6];101(12):2924–30. Available from: http://Insights.ovid.com/crossref?an=00007890-201712000-00019 [ Links ]
35. Zimmerhackl LB, Hofer J, Cortina G, Mark W, Würzner R, Jungraithmayr TC, et al. Prophylactic eculizumab after renal transplantation in atypical hemolytic–uremic syndrome. N Engl J Med [Internet]. 2010 May 6 [cited 2019 Aug 6];362(18):1746–8. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMc1001060 [ Links ]
36. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol [Internet]. 2010 Oct [cited 2019 Aug 6];5(10):1844–59. Available from: http://cjasn.asnjournals.org/lookup/doi/10.2215/CJN.02210310 [ Links ]
37. Gale DP, Pickering MC. Regulating complement in the kidney: insights from CFHR5 nephropathy. Dis Model Mech [Internet]. 2011 Nov 1 [cited 2019 Aug 6];4(6):721–6. Available from: http://dmm.biologists.org/cgi/doi/10.1242/dmm.008052 [ Links ]
Hugo Fernandes Diniz
Serviço de Nefrologia, Centro Hospitalar e Universitário de São João,
Alameda Prof. Hernâni Monteiro, 4200-319 Porto
E-mail: hugo.rc.dz@gmail.com
Disclosure of potential conflicts of interest: none declared
Received for publication: Aug 24, 2019
Accepted in revised form: Oct 20, 2019


{kind=link}
{kind=link}