










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.33 no.4 Lisboa dez. 2019
https://doi.org/10.32932/pjnh.2020.01.051
CASE REPORT
Cryoglobulinemic HCV-related membranoproliferative glomerulonephritis with TMA – secondary HUS or infection-triggered aHUS?
Ana Catarina Teixeira1, Helena Pinto1,2, Nuno Afonso1,2, Fátima Costa1, Carol Marinho3, Vítor Sousa3, Jorge Pratas1, Rui Alves1,2
1 Serviço de Nefrologia, Centro Hospitalar Universitário de Coimbra
2 Clínica Universitária de Nefrologia da Faculdade de Medicina da Universidade de Coimbra
3 Serviço de Anatomia Patológica, Centro Hospitalar Universitário de Coimbra
ABSTRACT
Introduction: Membranoproliferative glomerulonephritis is a pattern of glomerular injury on renal biopsy with characteristic light microscopic changes, namely mesangial hypercellularity, endocapillary proliferation and double contour formation along the glomerular capillary walls. This pattern does not represent a disease per se but can occur as a result of different pathologic processes. Case Presentation: We present a case of a 37‑year‑old caucasian female patient with thrombotic microangiopathy (TMA), severe hypertension and immune‑complex‑mediated glomerulonephritis, secondary to hepatitis C virus (HCV) infection. Steroid therapy and fresh‑frozen plasma infusions were started, with progressive improvement of lab results. Treatment of HCV infection was started with direct‑acting antiviral therapy. The results of the genetic study of the complement alternative pathway regulatory proteins were positive for a homozygote variant of the gene CFHR5 (p.Val110Ala) and a homozygote deletion on CFHR3/CFHR1 proteins. Discussion: Hypertension is commonly associated with TMA and could explain the features of this case. Nevertheless, the differential diagnosis is often difficult because TMA caused by hypertension is indistinguishable from all other entities. The finding of a homozygote variant of the gene CFHR5, and a homozygote deletion on CFHR3/CFHR1 proteins, raised the possibility that this case might have been an Atypical Hemolytic Uremic Syndrome (aHUS) triggered by the endothelial damage caused by the HCV‑related cryoglobulinemic glomerulonephritis.
Keywords: Atypical Hemolytic Uremic Syndrome, Complement system, Membranoproliferative glomerulonephritis, Thrombotic microangiopathy
INTRODUCTION
Membranoproliferative glomerulonephritis (MPGN) is a pattern of glomerular injury on renal biopsy with characteristic light microscopic changes, namely mesangial hypercellularity, endocapillary proliferation and double contour formation along the glomerular capillary walls.1
This pattern does not represent a disease per se but can occur as a result of different pathologic processes. The histological changes result from the deposition of immunoglobulins, complement factors, or both in the glomerular mesangium and along the glomerular capillary walls.2,3
Advances in the understanding of the pathogenesis of MPGN have resulted in a new classification of MPGN based on pathogenetic pathways.
In this system, MPGN is nowadays classified as being mediated by immune complexes, by complement dysregulation or rarely by mechanisms not involving immunoglobulin or complement deposition, such as endothelial injury. Autoimmune diseases, monoclonal gammopathy/dysproteinemias, and chronic infections are common causes of immune‑complex‑mediated MPGN.4
The clinical presentation is diverse, with variable degrees of kidney impairment; hypertension may or may not be present. The diagnosis is made by renal biopsy.2
The simultaneous presence of multiple diseases in a single host is rare, but occasionally multiple diseases may contribute to renal injury and the diagnosis and appropriated treatment may be a challenge.
We present the case of a 37‑year‑old female caucasian patient, who, in association to what it seemed an MPGN‑associated with cryoglobulinemia and HCV infection, also presented with severe thrombotic microangiopathy. In association she had a positive complement alternative pathway genetic study with a pathogenic mutation associated with the development of anti‑factor H autoantibodies. This raised the hypothesis of a possible complement role in this case and plunged us into the difficult differential diagnosis of atypical aHUS versus secondary thrombotic microangiopathy.
CASE PRESENTATION
A 37‑year‑old female Caucasian patient presented to the emergency department with a history of intense asthenia, anorexia and weight loss (30 Kg within three months). She had no other complaints, was under no medication and did not take any illicit drugs. There was no recent history of infection.
She had a history of poorly controlled hypertension with an erratic adherence to a prescription of amlodipine plus valsartan. Past medical history also included 11 pregnancies, with eclampsia complicating the last one, leading to miscarriage.
On physical examination severe hypertension (210/140mmHg) was present. An ophthalmologic evaluation was performed and revealed retinal lesions secondary to chronic hypertension (arteriolar narrowing) and macular degeneracy of the retina.
Findings on cardiovascular and respiratory examinations were noncontributory. The abdomen was soft, non‑distended, and nontender without masses or detectable organomegaly. Petechial lesions were apparent on the lower limbs and there was slight peripheral edema.
Initial lab results showed an acute kidney injury (serum creatinine 150 μmol/L; previous serum creatinine was 104 μmol/L); urinalysis demonstrated the presence of hematuria (15 red blood cells per high power field) and proteinuria (70mg/dL). Additionally, there was acute hemolytic anemia (serum hemoglobin 6.6gr/dl, with 12% of reticulocytes, erythrocyte fragmentation on peripheral blood smear and lowered serum haptoglobin, <0.07gr/dl), with thrombocytopenia (116000). Other relevant findings included high lactate dehydrogenase (LDH) (3376UI/L) and slight hyperbilirubinemia (64 μmol/L). The abdominal ultrasound showed a liver with heterogeneous echotexture and an enlarged spleen. The renal ultrasound reveled hyperreflectivity of the renal parenchyma with no other abnormalities. She was admitted to the nephrology ward for further treatment and evaluation. Intensive anti‑hypertensive treatment was started with subsequent normalization of blood pressure.
Additional investigations were performed including negative Coombs test; normal ADAMTS13 activity (99 %); negative pregnancy test; very low C4 (0.04 g/L, normal range 0.10 – 0.40) and C3 slightly decreased (0.85 g/L, normal range 0.90 – 1.80); positive HCV antibodies with active viral replication (5.77 log), genotype 1b, and positive cryoglobulins. All other immune studies (ANA, ANCA, dsDNA, ENA) were negative, as were HBV and HIV testing. Despite adequate control of hypertension, the hemolysis persisted after 3 days of hospitalization.
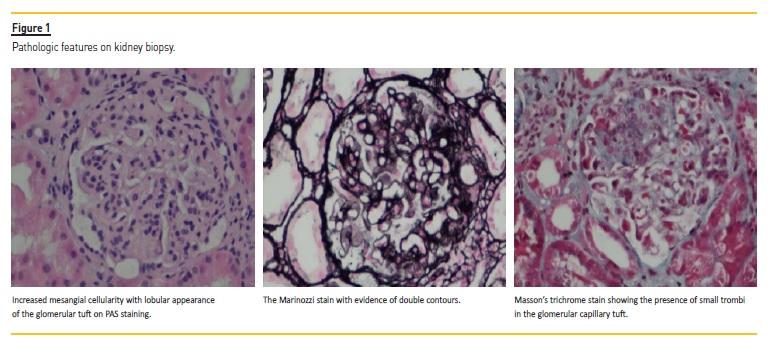
A renal biopsy was performed. The biopsy contained 9 glomeruli, one with global sclerosis. The remaining glomeruli showed mesangial hypercellularity and thickening of the glomerular capillary wall. Marinozzis technique highlighted the presence of double contours. About 25% tubular atrophy was also present. Massons trichrome stain showed the presence of small thrombi in the glomerular capillary tuft.
The immunofluorescence contained 12 glomeruli for analysis and showed positivity for mesangial granular deposition of IgM (++) and C3 (++).
The features described are compatible with membranoproliferative glomerulonephritis with positive IgM and C3 mesangial granular deposition on immunofluorescence.
Steroid therapy was initiated (3 IV pulses of 500mg prednisolone followed by oral prednisolone 1mg/Kg/day) and fresh‑frozen plasma infusions were started with progressive improvement of hematological lab results. There was a slight worsening in renal function during the hemolysis, with a maximum serum creatinine value of 180 μmol/L.
As hemolysis improved, renal function stabilized at creatinine values around 150 μmol/L. Plasma therapy was suspended after 20 days. Treatment of HCV infection was started with direct‑acting antiviral therapy.Figure 1
The patient completed 12 weeks of antiviral therapy that was effective in eliminating viral replication. During this period, the hematological parameters were normal and serum creatinine remained stable, around 150umol/l. After completion of antiviral therapy, the patient abandoned all consultations and was lost to follow‑up.
Later on, the results of the genetic study of the complement alternative pathway (AP) regulatory proteins were made available and were positive for a homozygote variant of the gene CFHR5 (p.Val110Ala) described on the databases as pathogenic and a homozygote deletion on CFHR3/CFHR1 proteins. This deletion is associated with the development of anti‑factor H autoantibodies.
DISCUSSION
Our patient presented with Coombs negative hemolytic anemia, with schistocytes in the peripheral blood smear, thrombocytopenia and acute organ injury (in this case, acute kidney injury). This pattern has the clinical and laboratory hallmarks of acute TMA. TMA is a quickly evolving, life‑threatening condition and its prompt recognition is critical to initiate early treatment and so improve prognosis.5,6
Although TMAs pattern should be promptly identified in clinical practice, the determination of its cause is usually difficult and time‑consuming.
Thrombotic microangiopathies include a group of pathologic conditions that primarily show lesions in the microvasculature and typically represent two major subgroups: thrombotic thrombocytopenic purpura (its diagnosis is promptly excluded by determining ADAMTS13 levels) and hemolytic uremic syndrome (HUS). The term hemolytic uremic syndrome encompasses 3 different entities: typical HUS caused by a shiga toxin‑producing Escherichia coli; primary atypical HUS (aHUS) resulting from primary dysregulation of the complement alternative pathway, with pathogenic variants in the genes coding for proteins and regulators of the alternative pathway found in 40% to 60% of cases; and secondary HUS caused by a variety of etiologies whose common characteristic is to induce diffuse endothelial damage leading to TMA.7 This latter list of etiologies is extensive, including systemic infections (bacterial, viral and fungal), malignancies, drugs, autoimmune disorders, and even glomerulonephritis, making the differential diagnosis truly challenging.8 To make matters worse, central to the pathophysiology in many of these conditions, there is a second hit complement system activation that aggravates the TMA and can make it unresponsive to the treatment of the initial cause.7 This complement activation can also be the trigger for an aHUS. Indeed, in approximately two‑thirds of aHUS cases, a complement‑activating condition can also be found.6
We present a challenging case of a woman who presented initially with TMA, severe hypertension and immune‑complex‑mediated (probably cryoglobulinemic) glomerulonephritis, secondary to HCV infection.
Hypertension is commonly associated with TMA and could explain the features of this case. Nevertheless, the differential diagnosis is often difficult because TMA caused by hypertension is indistinguishable from all other entities.
The key to the differential diagnosis is the evolution of the clinical condition: TMA secondary to hypertension should improve quickly with blood pressure control.5 In this patient, although prompt antihypertensive treatment and blood pressure control was achieved, the hemolysis persisted, and so other hypotheses emerged.
The presence of consumption of complement´s classical pathway (very low level of C4 with slightly decreased C3) with positive cryoglobulins and the fact that the patient had eleven previous pregnancies (an important trigger for aHUS), without developing TMA, made us conclude that, in this case, HUS was probably secondary to glomerular endothelial damage associated with the cryoglobulinemia; hence the
MPGN in itself. Secondary forms of HUS are heterogeneous and have distinct mechanisms of initial endothelial cell injury but share a common final phenotype of activated and prothrombotic endothelial cells. Transient complement activation may occur in some patients with secondary HUS and thus promote the TMA process.9
This interpretation of the case determined the therapeutic options. The presence of TMA made us start plasma therapy, but the presence of MPGN with AKI determined the use of steroid therapy and the start of antiviral therapy. Although we fully recognize the potential advantages of plasma exchange vis‑a‑vis plasma infusion in relation to the total volume of plasma infused and the potential for removing complement activating factors (e.g. anti‑factor H antibodies), the fact remains that, in the largest analysis available by Noris et al, there were no outcome differences between the therapeutic options.10 In our case, logistical constraints, the fact that the patient had preserved diuresis, and the conviction of an immune‑mediated process, led us to the decision of plasma infusion.
Later, the results of the study of the genes regulating the complements alternate pathway, and the finding of a homozygote variant of the gene CFHR5 (p.Val110Ala), and a homozygote deletion on CFHR3/CFHR1 proteins, raised the possibility that this case might have been an aHUS triggered by the endothelial damage caused by the HCV‑related cryoglobulinemic glomerulonephritis.
The finding of rare genetic variants in complement genes must be interpreted with great caution in cases of TMA that have a clear and plausible secondary cause. Indeed, a recent analysis of a French cohort of 110 patients with secondary HUS found that the frequency of complement gene rare variants was similar in patients with secondary HUS (5%) and in healthy individuals (6% and 8% in French and European controls, respectively).8 This contrasts with atypical HUS, which has been linked in a significant number of large studies to a high incidence (40%–70%) of rare and mostly pathogenic complement genes variants and thus is assumed to be mediated by uncontrolled complement activation.8
The homozygote variant of the CFHR5 gene found in our patient is a rare variant described in European databases (MAF 0.00012); its significance is uncertain and, with an in silico score of 3, is considered to be likely pathogenic. The CFHR5 protein is a surface‑acting complement activator and variations in the CFHR5 gene are linked to CFHR glomerulonephritis.11 The CFHR5 protein binds to laminin‑521, the major constituent of the glomerular basement membrane, and to mesangial laminin‑211, fostering complement activation at damaged sites.11 It is thus conceivable that a pathogenic variant of this protein could potentiate complement activation in an endothelial cell initially damaged by an immune complex process, leading to the subsequent development of complement‑mediated TMA.
The homozygote deletion on CFHR3/CFHR1 protein is associated with the development of anti‑factor H autoantibodies and its pathogenic effect is dependent on the presence of these antibodies, that block the complement regulatory effect of factor H.12 Unfortunately we were not able to perform an assay to determine if these antibodies were present.
In aHUS, a genetic or acquired defect in the regulators of complement activation may lead to unchecked spontaneous activation of the AP. Occasionally, the defect is not severe enough to cause dysregulation of the complement system under normal conditions and a trigger (for example, infection, vaccinations, immunosuppressive or antineoplastic drugs, oral contraceptives, pregnancy) is necessary to induce the overactivation of the AP.13
We think that in this case, the TMA was caused primarily from capillary endothelial lesion caused by HCV infection with cryoglobulinemia and subsequent MPGN, this being the trigger that disrupted the fragile balance between activation and restraint, thereby instigating activation of the AP, leading to HUS. Heat‑insoluble cryoglobulins have been demonstrated as toxic to endothelial cells in vitro, and such an effect in vivo may promote the microangiopathic process.14
HCV stimulates the production of a variety of autoantibodies. These autoantibodies produced by cryoglobulinemia‑induced B‑cell lymphoproliferation may also have a thrombogenic potential (e.g. antiplatelet antibodies, which could cause thrombocytopenia and endothelial injury).14,15
Some studies have suggested that atypical HUS and secondary HUS have no common genetic risk factors and that the presentation and outcome are different.8
Our case, however, fully illustrates the thin line separating these entities and the difficulties in performing a differential diagnosis between them. Our patient, despite having a clear secondary cause for the TMA, also has a genetic study of the genes codifying the regulatory proteins of complements AP that raised the question of an eventual role of a primary complement AP deregulation on the pathophysiology of the case. All things considered, however, we think that, in our case, the TMA was most probably caused by the HCV‑related cryoglobulinemic MPGN and that the genetic findings described, although possibly contributory, are less relevant in the pathophysiology of our case.
Treatment of this type of TMA includes first and foremost the withdrawal or treatment of the underlying disease and thus the removal or control of the initial main trigger of TMA, in this case, the HCV infection treatment, and treatment of the acute glomerulonephritis with steroids.9 The hypothesis of associated complement activation on the pathophysiology justifies the use of plasma therapy and could, in cases of persistence of the TMA, also raise the possibility of using complement blockade therapy.7
References
1. Fervenza FC, Sethi S. Clinical presentation, classification, and causes of membranoproliferative glomerulonephritis. UptoDate [Intranet] [Internet]. 2014;1–11. Available from: http://www.uptodate.com/contents/clinical‑presentation‑classification‑and‑causes‑of‑membranoproliferative‑glomerulonephritis?source=search_result&search=glomerulonephritis&selectedTitle=5~150
2. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis – a new look at an old entity. N Engl J Med [Internet]. 2012;366(12):1119–31. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMra1108178 [ Links ]
3. Sethi S, Fervenza FC. Pathology of renal diseases associated with dysfunction of the alternative pathway of complement: C3 glomerulopathy and atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. 2014;40(4):416–21. [ Links ]
4. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: pathogenetic heterogeneity and proposal for a new cassification. Semin Nephrol [Internet]. 2011;31(4):341–8. Available from: http://dx.doi.org/10.1016/j.semnephrol.2011.06.005 [ Links ]
5. Azevedo A, Faria B, Teixeira C, Carvalho F, Neto G, Santos J, et al. Portuguese consensus document statement in diagnostic and management of atypical hemolytic uremic syndrome. Port J Nephrol Hypertens. 2018;32(October):211–32. [ Links ]
6. Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia [Internet]. 2015;35(5):421–47. Available from: http://dx.doi.org/10.1016/j.nefroe.2015.11.006. [ Links ]
7. Caravaca‑Fontan F, Praga M. Complement inhibitors are useful in secondary hemolytic uremic syndromes. Kidney Int [Internet]. 2019 Oct 1;96(4):826–9. Available from: https://doi.org/10.1016/j.kint.2019.07.006 [ Links ]
8. George JN, Charania RS. Evaluation of patients with microangiopathic hemolytic anemia and thrombocytopenia importance of diagnosing microangiopathic hemolytic anemia for decisions on therapy. Semin Thromb Hemost. 2013;39(212):153–60. [ Links ]
9. Le Clech A, Simon‑Tillaux N, Provôt F, Delmas Y, Vieira‑Martins P, Limou S, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019; 95 (6): 1443 – 1452; [ Links ]
10. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844–59. [ Links ]
11. Rudnick RB, Chen Q, Stea ED, Hartmann A, Papac‑Milicevic N, Person F, et al. FHR5 binds to laminins, uses separate C3b and surface‑binding sites, and activates complement on malondialdehyde‑acetaldehyde surfaces. J Immunol. 2018;200(7):2280–90. [ Links ]
12. Oliveira N, Neto G, Azevedo A, Santos J, Fidalgo T. 50 Perguntas chave na nefrologia – no diagnóstico e terapêutica das microangiopatias trombóticas. Permanyer Portugal; 2019; 15–24. [ Links ]
13. De Vriese AS, Sethi S, Van Praet J, Nath KA, Fervenza FC. Kidney disease caused by dysregulation of the complement alternative pathway: an etiologic approach. J Am Soc Nephrol [Internet]. 2015;26(12):2917–29. Available from: http://www.jasn.org/cgi/doi/10.1681/ASN.2015020184 [ Links ]
14. Hao Wu, MS, Hong‑Bin Zou, MS, Ying Xu, MD, Li Zhang, MD, Yu‑Jun Du, MD, Jin‑Yu Yu, MS, Rong‑Hua Lu, MS and Xiang Li M. Hepatitis C virus‑related heat‑insoluble cryoglobulinemia. Am J Med Sci. 2013;346(4):345–8. [ Links ]
Ana Catarina Teixeira
Nephrology Department, Centro Hospitalar Universitário de Coimbra
Hospitais da Universidade de Coimbra, Praceta Professor Mota Pinto, 3000‑075 Coimbra, Portugal.
E‑mail: acteixeira.ct@gmail.com
Disclosure of potential conflicts of interest: none declared
Received for publication: Oct 1, 2019
Accepted in revised form: Nov 27, 2019


{kind=link}