










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.34 no.1 Lisboa mar. 2020
https://doi.org/10.32932/pjnh.2020.04.063
CASE REPORT
Can cryoglobulinemia trigger ANCA vasculitis?
Ivo Cunha1, M. Teresa Santos2, P. Ricardo Pereira1
1 Internal Medicine Department; Hospital Pedro Hispano
2 Nephrology Department; Hospital Pedro Hispano
ABSTRACT
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis is a rare and heterogeneous group of autoimmune diseases. These pauciimmune vasculitis differ from the cryoglobulinemic vasculitis mediated by immune complexes. We report the case of a 79-year-old male with silicosis and chronic alcoholic liver disease, presenting with a rapidly progressive glomerulonephritis and pancytopenia. Work-up revealed hypocomplementemia, polyclonal hypergammaglobulinemia, type 3 cryoglobulinemia, p-ANCA positivity and elevated anti-MPO. Renal biopsy showed a pattern of chronic interstitial nephritis with fibrocellular crescents, and immunofluorescence staining was negative. Treatment was started with corticosteroids and rituximab with improvement of the renal function, decrease of the anti-MPO titer and disappearance of the cryoglobulinemia. In this case, renal injury was caused by ANCA vasculitis, whose etiology remains unknown despite the recognizable risk factor for ANCA formation (silicosis). The importance of the cryoglobulinemia is not clear, as it could be part of the pathogenesis or just an epiphenomenon secondary to the autoimmune and the chronic liver diseases.
Keywords: Crescents, cryoglobulinemia, interstitial nephritis, rapidly progressive glomerulonephritis, vasculitis.
INTRODUCTION
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis is a set of rare autoimmune diseases that predominantly affect small blood vessels, in the majority of cases with fibrinoid necrosis phenomena, that lead to the destruction and thrombosis of the involved vessels1. They comprise microscopic polyangiitis, granulomatous polyangiitis, eosinophilic granulomatous polyangiitis, and ANCA vasculitis limited to the kidney, seen by many authors as a microscopic or granulomatous polyangiitis without systemic expression2,3. However, current theories suggest they may be one sole disease4.
Several risk factors have been associated with the formation of ANCAs, in a genetically susceptible individual, namely exposure to viral or bacterial infections such as Staphylococcus aureus; exposure to specific types of climate; exposure to drugs such as propylthiouracil and exposure to silica dust5. Regarding this latter risk factor, its association with the formation of ANCAs, mainly anti-myeloperoxidase (MPO), is now known, although there are no conclusive studies that relate the exposure to the establishment of the systemic vasculitis6.
There seems to be a connection between environmental and genetic factors, based on molecular mimicry as postulated by the new currents of thought such as the theory of complementarity of auto-antigens7, creating the perfect environment for the development of the vasculitis5.
Clinically, their presentation varies from nonspecific constitutional symptoms to neurological manifestations such as peripheral neuropathy; pulmonary and upper airway disease; cutaneous changes, namely purpura; and other less common manifestations such as cardiomyopathy, venous thrombosis or orbital tumor3. The renal involvement is almost the rule in ANCA vasculitis, especially in microscopic polyangiitis and granulomatous polyangiitis. In most cases, it occurs in the form of a rapidly progressive glomerulonephritis or subacute or chronic interstitial nephritis8, with renal biopsy crucial for the diagnosis and definition of prognosis. Because of the rarity and heterogeneity of this type of vasculitis, its treatment is challenging, based on systemic immunosuppression, without which prognosis is poor3.
Cryoglobulins are immunoglobulins that precipitate in vitro at temperatures below 37ºC, causing symptoms due to hyperviscosity and autoimmune mechanisms, namely vasculitic. Cryoglobulinemia is classified essentially into two subgroups. Type I is associated with hematological diseases predominantly of B-lineage such as multiple myeloma and Waldenstrom macroglobulinemia, with cryoglobulins being composed of monoclonal immunoglobulins, typically IgG or IgM and, in rare cases, IgA. In type II/III, cryoglobulins have a mixed composition, consisting of monoclonal IgM immunoglobulins with rheumatoid factor activity and polyclonal IgG immunoglobulins. This type is particularly associated with hepatitis C virus infection, but also with other infections such as HIV or hepatitis B, as well as other chronic liver, autoimmune and lymphoproliferative diseases. Sometimes, there is no cause for cryoglobulinemia and patients can remain asymptomatic9,10.
When there is clinical expression, cutaneous purpura is the main manifestation and can be part of the Meltzer triad when it occurs simultaneously with arthralgias and myalgias. The peripheral nerves may also be affected in the form of multiplex mononeuritis. The renal system is mainly affected in the form of type 1 membranoproliferative glomerulonephritis and the digestive system in the form of intestinal vasculitis. Blood work usually reveals hypocomplementemia, sometimes only of C4, and increased rheumatoid factor activity. In addition it is also important to quantify and determine the type of cryoglobulins.
The prognosis is variable according to the underlying disease and the degree of the involvement of the affected organs. The treatment comprises the support of the affected organs and mainly the treatment of the underlying disease responsible for the development of cryoglobulins and cryoglobulinemia9,10.
CLINICAL CASE
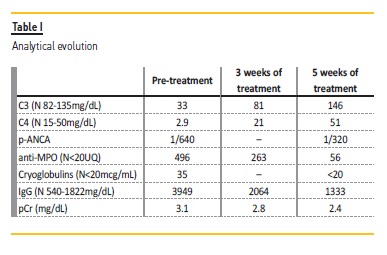
We present the clinical case of a 79-year-old man with chronic alcoholic liver disease (Child-Pugh A), pulmonary silicosis, hypocoagulated due to atrial fibrillation and vascular risk factors, namely chronic hypertension, controlled with only one drug, and dyslipidemia. An ultrasound pattern suggestive of diffuse hepatic steatosis was present, as well as a mild splenic enlargement. He had a moderate thrombocytopenia (99000exp3/uL), hyperferritinemia (1143ng/mL) with transferrin saturation of 27% and elevated GGT (153U/L). Serum protein electrophoresis revealed a beta-gamma bridging, associated with polyclonal hypergammaglobulinemia and hypocomplementemia of C3 (33mg/dL) and C4(2.9mg/dL) was also present. In addition to the decrease in both complement fractions, which was initially assumed to be associated with liver disease, there were no other indicators of hepatic impairment (total bilirubin 0.9mg/dL, albumin 3.9g/dL and fibrinogen 4.24g/L).
Autoimmune causes for liver disease were excluded. On objective examination, there were no major alterations such as purpuric lesions, cutaneous ulcers, signs of arthritis, Raynaud's phenomena or digital lesions, nor clinical manifestations suggestive of peripheral neuropathy. In the first evaluation, he had a plasma creatinine (pCr) of 1.2mg/dL, with a glomerular filtration rate (GFR) of 62.08mL/min/1.73m2 MDRD-4, and a summary urine test with no detectable changes.
During follow-up, about two years later the patient developed a rapidly progressive renal lesion with no obstructive component, with a maximum pCr of 3.1mg/dL, microscopic hematuria and non-nephrotic proteinuria (protein/creatinine ratio of 500mg/g and albumin/creatinine ratio of 73.58mg/g), without erythrocyte dysmorphia nor erythrocyte cylinders in the urine sediment. A concomitant pancytopenia (hemoglobin 11.1g/dL, leukopenia 3.10exp3/uL, and worsening thrombocytopenia 87000exp3/uL) developed. Further investigations revealed a p-ANCA titer of 1/640 and an anti-MPO of 496 UQ (N <20), a sedimentation rate (ERS) of 112mm/1h, a rheumatoid factor of 12U/mL (N<30U/mL), a polyclonal hypergammaglobulinemia (with an IgG increase from 2230mg/dL to 3949mg/dL, with no IgG4 predominance) and serum cryoglobulins of IgG and IgA, with polyclonal characteristics, of 35mcg/mL (N<20) without rheumatoid factor nor complement activity. Viral serologies were negative, namely for hepatitis C virus.
A myelogram revealed only hypocellular bone marrow without changes in maturation or malignancy, and immunophenotyping of peripheral blood and thoracoabdominal-pelvic CT showed no evidence of lymphoma. Serum and urinary immunofixation were also negative. Renal biopsy was performed and showed the presence of 18 glomeruli, five of them with global sclerosis and two with fibrocellular crescents. The interstitial compartment showed 50% of interstitial fibrosis and tubular atrophy and reactive lymphocyte infiltration (mainly CD3+) and arterioles with hyalinosis (Figure 1, 2, 3, 4). Immunofluorescence staining was negative for IgA, IgG, IgM, C3 and C1q. Immunohistochemistry with anti-CD3 serum showed the presence of reactive lymphocytes CD3+.
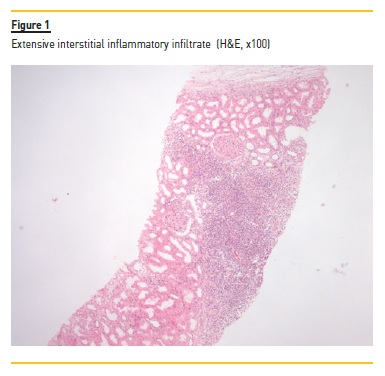
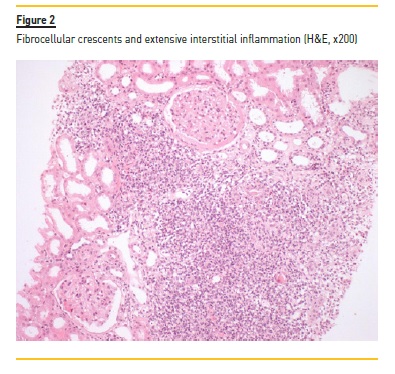
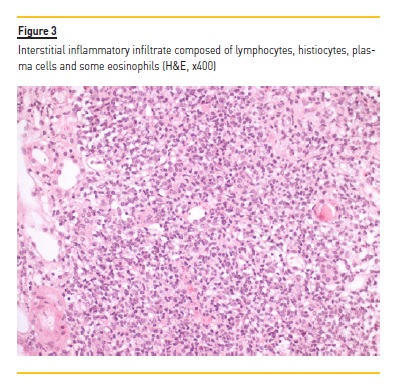
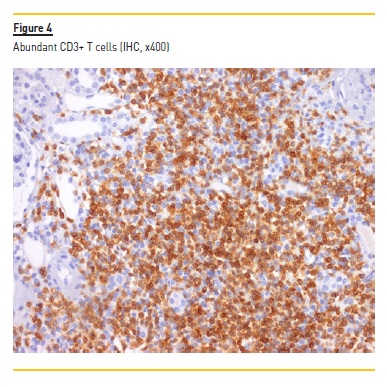
Induction treatment was started, based on prednisolone 60mg/day with progressive weaning up to 2.5mg/day and rituximab 375mg/m2 weekly for 4 weeks. The treatment resulted in improvement (Table 1) of the pancytopenia; normalization of the C3 (146mg/dL) and the C4 (51mg/dL) values; normalization of the IgG values; and disappearance of the cryoglobulinemia (<20mcg/mL). The anti-MPO (56UQ) and p-ANCA (1/320) titers were also reduced, and a partial recovery of the renal function was observed, even though, with a progression to chronic stage 4/A2.
DISCUSSION
The association between the presence of ANCAs and cryoglobulinemia at the same clinical time has been previously reported with renal biopsy showing cryoglobulinemic lesions and presumption of ANCAs as false positives11. What we now report with this clinical case is the opposite, that is, the diagnosis in renal biopsy of an ANCA vasculitis in a patient that presented initially with hypocomplementemia and polyclonal hypergammaglobulinemia attributed to cryoglobulinemia.
Although hypocomplementemia and polyclonal hypergammaglobulinemia were initially assumed to be secondary to the chronic liver disease, despite being disproportionate to the degree of hepatic impairment, the patient presented no other clinical or laboratorial evidence suggestive of cryoglobulinemia.
The diagnosis of ANCA vasculitis was supported by the demonstration in the renal biopsy of a pattern of chronic interstitial nephritis with presence of reactive lymphoid follicles with inflammatory vascular lesions, fibrocellular crescents and arteriolitis. These changes were compatible with a pauci-immune vasculitis with a predominant tubulointerstitial involvement. This is a common expression of a systemic vasculitis and has a strong association with circulating anti-MPO antibodies4. Also in favor of this diagnosis is the clinical presentation in the form of a rapidly progressive renal lesion, which is one of the typical forms of presentation in ANCA-MPO vasculitis8. The association with pulmonary silicosis is also important, which, in addition to being a known risk factor for renal damage and progression to chronic kidney disease,6,12, is also a known risk factor for the formation of anti-MPO antibodies. However, the mechanism by which these antibodies become pathogenic with subsequent development of vasculitis remains unclear5.
What made the presentation atypical for a singular diagnosis of ANCA vasculitis was the identification of hypocomplementemia and the long-standing polyclonal hypergammaglobulinemia preceding the acute renal failure, which disappeared in the first case and was significantly reduced in the second, after treatment completion. This may suggest that the patient already had circulating cryoglobulins although asymptomatically and without evidence of renal involvement.
On the other hand, the immunofluorescence was negative and the immunohistochemistry was positive for reactive lymphocytes CD3+, which favors a ANCA vasculitis. In this particular case, chronic liver disease may have been the main etiology for the polyclonal hypergammaglobulinemia13, thus creating conditions for the benign proliferation of B cells and the consequent development of a cryoglobulinemia in a patient with, at least, environmental predisposition (silicosis) for the development of ANCAs.
Even though this could be the case of two distinct entities in the same patient, it is still possible that some type of interaction between cryoglobulins and ANCAs occurs. The association between ANCAs and cryoglobulins has been questioned14,15 without any clear definition of their possible interaction. What seems most likely, given the basic constitution of both immunoglobulins and the efficacy of rituximab in its treatment, is a possible role of B lymphocytes in the regulation or deregulation of the interaction between the two types of antibodies.
But the question remains whether the quiescent cryoglobulins in the patient in question will have contributed and could eventually be identified as a risk factor for the development of ANCAs and later for the evolution to a vasculitic process. With regard to the evolution to a chronic renal disease, there were multiple contributions in this patient, from chronic hypertension, to ANCA vasculitis with progression to chronic interstitial nephritis and the contribution of silicosis itself.
Finally, with regard to the renal biopsy, in this case it was crucial as the only way to understand and give a name to the disease.
CONCLUSION
This case shows the role of known risk factors for the appearance of ANCA antibodies, such as silicosis, and raises questions about the contribution of other factors, namely cryoglobulins, highlighting the possible role of the B lymphocytes and the mechanisms that confer pathogenicity to the immunoglobulins they produce. Even though some theories such as the theory of complementarity of autoantigens help to understand the mechanisms of autoimmunity and its triggering factors, it is difficult to understand the true nature of ANCAs. In addition, there are still many doubts about what is necessary for their connection to neutrophils and monocytes and what mechanisms lead to the process of vasculitis.
References
1. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. [ Links ]
2. Yates M, Watts R. ANCA-associated vasculitis. Clinical Medicine, 2017;17(1): 60-64. [ Links ]
3. Pagnoux C. Updates in ANCA-associated vasculitis. Eur J Rheumatol. 2016;3:122-133. [ Links ]
4. Binda V, et al. ANCA-associated vasculitis with renal involvement. J Nephrol. 2018;31(2):197-208. [ Links ]
5. de Lind van Wijngaarden RAF: Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody–associated vasculitis: the cause Is hidden, but the result is known. Clin J Am Soc Nephrol. 2008;3:237-252. [ Links ]
6. Melissa L, et al. Silicosis and chronic renal disease. Am J Ind Med. 2015;58:730-736. [ Links ]
7. Pendergraft III WF, et al. Autoimmunity is triggered by cPR-3(105–201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10(1):72-79. [ Links ]
8. Syed R, et al. Pauci-immune crescentic glomerulonephritis: an ANCA-associated vasculitis. BioMed Research International Volume 2015 (2015), Article ID 402826. [ Links ]
9. Ramos-Casals M, et al. The cryoglobulinaemias. Lancet. 2012;379:348–360. [ Links ]
10. Muchtar E, et al. How I treat cryoglobulinemia. Blood. 2017;129(3):289-298. [ Links ]
11. Lamprecht P, et al. Mixed cryoglobulinaemia, glomerulonephritis, and ANCA: essential cryoglobulinaemic vasculitis or ANCA-associated vasculitis? Nephrol Dial Transplant. 1998;13:213-221. [ Links ]
12. Rosenman KD, et al. Kidney disease and silicosis. Nephron. 2000;85:14-19. [ Links ]
13. Dispenzieri A, et al. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc. 2001;76:476-487. [ Links ]
14. Ferri C, et al. B-cells and mixed cryoglobulinemia. Autoimmunity Reviews. 2007;7:114-120. [ Links ]
15. Lamprecht P, et al. Prevalence of ANCA in mixed cryoglobulinemia and chronic hepatitis C virus infection. Clin Exp Rheumatol. 2003;21(32):S89-94. [ Links ]
Ivo Cunha, MD
Nephrology Department; Hospital Pedro Hispano
E -mail: ibooo1@hotmail.com
Disclosure of potential conflicts of interest:none declared
Received for publication: Oct 20, 2019
Accepted in revised form: Feb 8, 2020

