










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.34 no.1 Lisboa mar. 2020
https://doi.org/10.32932/pjnh.2020.04.067
NEPHROPATHOLOGY QUIZ
Anemia, thrombocytopenia and acute kidney injury – a diagnostic challenge
Sara Cardoso Fernandes1, Ana Carina Ferreira1,2, Mário Góis1,3, Helena Viana1,3, Fernando Nolasco1,2
1 Nephrology Department, Hospital de Curry Cabral, Centro Hospitalar Universitário de Lisboa Central
2 Nova Medical School, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, Lisbon, Portugal
3 Laboratory of Renal Morphology, Nephrology Department, Hospital Curry Cabral – Centro Hospitalar Universitário de Lisboa Central
CLINICAL PRESENTATION
The authors present the case of a 72-year-old female with previous medical history of hypertension, ischemic cardiomyopathy and cerebrovascular disease with an ill-defined episode that resulted in brachial palsy.
The patient was followed in hematology clinic since 2014 for anemia, initially assumed as iron deficient, and thrombocytopenia, without leukopenia. She was referred to nephrology to investigate a fall in hemoglobin levels and platelet count, accompanied by acute kidney injury. Laboratory tests showed microcytic hypochromic anemia (Hb 10.4 g/dL), thrombocytopenia (75 000/uL platelets), prolonged activated partial thromboplastin time (aPTT 51.9 s) and worsening renal function (urea 109 mg/dL; creatinine 1.91 mg/dL; eGFR 26 ml/min/1.73m2).
The initial diagnostic workup revealed negative serology tests for B and C hepatitis and HIV infection; normal folate and vitamin B12 levels and low ferritin; absence of schistocytes in the blood smear; normal serum protein electrophoresis, serum immunofixation without a well-defined monoclonal spike, increased serum free kappa and lambda light chains without abnormal ratio and negative Bence Jones protein. Although the patient had slight elevated LDH levels, haptoglobin was within the normal range and ADAMTS13 activity levels were normal. We found a positive direct Coombs test, low C3 and C4 levels, positive antinuclear (ANAs), anticardiolipin and anti-beta2-glycoprotein antibodies as well as positive lupus anticoagulant. Anti-double-stranded DNA (anti-dsDNA) was negative.
Prolonged aPTT normalized after introducing a correction with phospholipids. Since deterioration of renal function persisted, a renal biopsy was performed despite unremarkable urine sediment.
QUESTIONS
1. Considering clinical history and laboratory results, what is the most likely diagnosis?
2. Given the histology findings, what is the definitive diagnosis?
3. What are the most common renal manifestations of this condition?
4. What is the appropriate treatment for this patient?
ANSWERS
1. Considering clinical history and laboratory results, what is the most likely diagnosis?
In the presence of anemia, thrombocytopenia and renal dysfunction, several systemic disorders should be excluded, namely infections(the most frequent ones being HIV and HCV), dysproteinemias or other bone marrow disorders, thrombotic microangiopathies (TMA) and autoimmune disorders. Serology tests for HIV and HCV were negative, excluding those from our list of possibilities. A monoclonal gammopathy was excluded, as we found no presence of a monoclonal immunoglobulin or its components. The presence of microangiopathic haemolytic anemia (MAHA) was ruled out in the absence of schistocytes, normal haptoglobin serum levels and positive direct Coombs test. The exclusion of MAHA allowed us to reject the hypothesis of TMA, which was also supported by normal ADAMTS13 activity levels.
Positive ANAs and hypocomplementemia suggested autoimmune disease with complement classical pathway activation, leading to antidsDNAS and antiphospholipid antibodies testing. The results showed triple positivity (positive lupus anticoagulant, anticardiolipin and antibeta2-glycoprotein antibodies) for antiphospholipid antibodies. A prolonged aPTT also supported the diagnosis of Antiphospholipid Syndrome (APS).
In order to confirm the diagnosis of APS, patients should meet at least one clinical criteria (one or more episodes of venous, arterial, or small vessel thrombosis in any tissue or organ, with unequivocal imaging or histology evidence of thrombosis or pregnancy morbidity) nd one laboratory criteria (presence of one or more of the abovementioned antiphospholipid antibodies on two or more occasions at least 12 weeks apart). Despite meeting the laboratory criteria, our patient had never been pregnant and the etiology of her previous neurologic event was not completely clarified1. We hoped to find a vessel thrombosis in renal biopsy in order to meet all the criteria.
2. Given the histology findings, what is the definitive diagnosis?
The renal biopsy consisted of renal capsule and cortical, and contained 21 glomeruli and medium-sized arteries. In light microscopy, an area with interstitial fibrosis and tubular atrophy and some ischemic glomeruli was evident (Figure 1). Figure 2 describes a small thrombus in afferent arteriole (arrow), with ischemic changes of the glomerular tuff. In Figure 3, we can observe a glomerulus with endotheliosis of the afferent arteriole (*), near of an area of mesangiolysis (arrow). In Figure 4, there is fibrotic medial hyperplasia of the artery and the lumen is irregular due to a mural thrombus organization (*). There were no immune deposits in immunofluorescence.
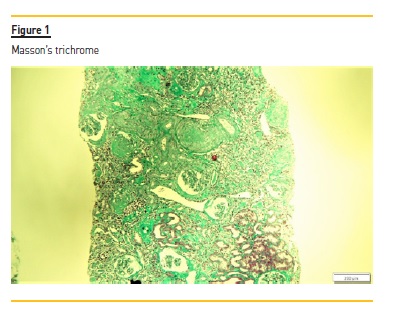
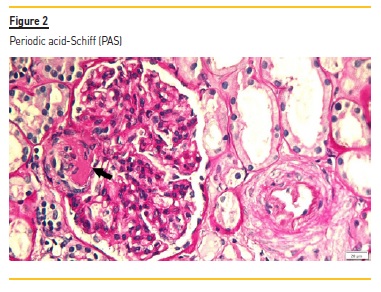
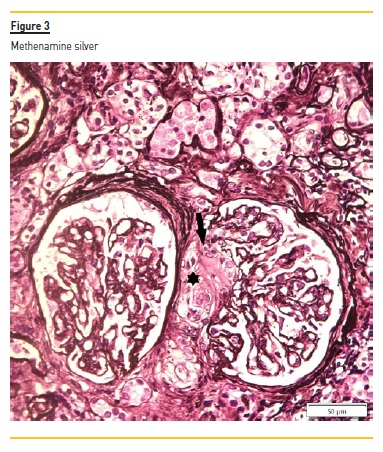
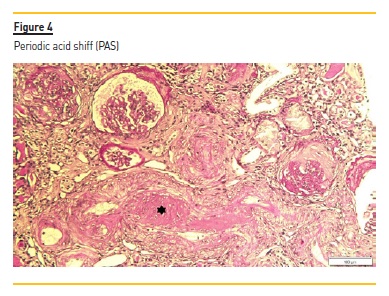
These clinical, histology and laboratory findings support the diagnosis of APS. This syndrome occurs either as a primary condition or in the setting of an underlying disease, usually systemic lupus erythematosus (SLE)2,3. Since our patient did not meet criteria for SLE or other autoimmune disease, APS was classified as primary.
3. What are the most common renal manifestations of this condition?
Renal involvement occurs in as many as 25 percent of patients with the primary APS. Renal manifestations in primary APS are primarily characterized by non-inflammatory occlusion of a broad spectrum of renal blood vessels, a finding that resembles thrombotic microangiopathy, as described above4. However, other glomerular lesions have been described in patients with primary APS, including minimal change disease, membranous nephropathy and pauci-immune glomerulonephritis5,6.
Patients with secondary APS related to SLE may present with microthrombi, characteristic of APS renal involvement, and immune complexes deposits, the hallmark of lupus nephritis7.
4. What is the appropriate treatment for this patient?
The mainstay of treatment for APS patients with history of thrombotic events is oral anticoagulation. Warfarin is the drug of choice, since direct oral anticoagulants showed a higher risk of recurrent thrombosis and are not routinely recommended8. In fact, triple positivity for antiphospholipid antibodies was associated with a 3.5-fold increased risk of recurrent thrombosis9. For most patients with APS, lifelong anticoagulation is necessary.
References
1. Ruiz-Irastorza G, Crowther M, Branch W et al. Antiphospholipid syndrome. Lancet 2010;376(9751):1498–1509. [ Links ]
2. Vianna JL, Khamashta MA, Ordi-Ros, J. et al. Comparison of the primary and secondary antiphospholipid syndrome: a European multicenter study of 114 patients. Am J Med 1994;96(3):3–9. [ Links ]
3. Cervera R, Serrano R, Pons-Estel, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis 2014; 74(6):1011-8.
4. Griffiths MH, Papadaki L, Neild GH. The renal pathology of primary antiphospholipid syndrome: a distinctive form of endothelial injury. Q J Med 2000;93:457–467. [ Links ]
5. Turrent-Carriles A, Herrera-Félix JP, Amigo M et al. Renal involvement in antiphospholipid syndrome. Front Immunol 2018;9(5):1–9. [ Links ]
6. Fakhouri F, Noël L-H, Zuber J. The expanding spectrum of renal diseases associated with antiphospholipid syndrome. Am J Kidney Dis 2003;41(6):1205–1211. [ Links ]
7. Tektonidou MG, Sotsiou F, Nakopoulou L et al. Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphospholipid antibodies. Arthritis Rheum 2004;50(8):2569–2579. [ Links ]
8. Keeling D, Mackie I, Moore GW et al. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012;157(02):47–58. [ Links ]
9. Dufrost V, Risse J, Zuily S et al. Direct oral anticoagulants use in antiphospholipid syndrome: are these drugs an effective and safe alternative to warfarin? A systematic review of the literature. Curr Rheumatol Rep 2016;18(74):3–8. [ Links ]
Mário Góis, MD
Laboratory of Renal Morphology, Hospital Curry Cabral
Centro Hospitalar e Universitário de Lisboa Central, Lisboa, Portugal
E -mail: mario.gois@chlc.min-saude.pt
Disclosure of potential conflicts of interest: none declared
Received for publication: Mar 31, 2020
Accepted in revised form: Apr 2, 2020

