











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Fabry disease (FD; OMIM 301500) is a rare X‑linked systemic disease caused by a GLA gene mutation. As a result, a deficiency of α‑galactosidase A leads to progressive accumulation of globotriaosylceramide (Gb3) and globotriaosylsphingosine (LysoGb3) in various organs, mostly in the vascular endothelium, heart and central nervous system.1,2Classic and more severe manifestations of FD are typically experienced by hemizygous males without residual enzyme activity.
These patients usually present FD characteristic symptoms such as neuropathic pain, cornea verticillata and angiokeratoma at a young age. Later in the course of the disease, hypertrophic cardiomyopathy, cardiac rhythm disturbances, progressive renal failure and stroke are frequent manifestations.3 Nonclassical FD, also called late‑onset FD, is characterized by a more variable disease course. Patients generally have residual enzyme activity and lower levels of LysoGb3, are less severely affected and disease manifestations are frequently limited to a single organ.4 In heterozygous females symptoms depend on the mutation and inactivation profile of the X chromosome (lyonization).
The spectrum of disease severity ranges from asymptomatic to a severe phenotype, similar to those of males with the classic phenotype.5-7
Diagnosis of FD is made by measuring α‑Gal A activity in plasma, leukocytes, dried blood spot or fibroblasts. A confirmative genotype analysis should be performed, especially in females since they can present a normal enzymatic activity.8,9The introduction of enzyme replacement therapy (ERT) changed the natural history of this disease by preventing glycosphingolipids deposition in the organs. Recognition of FD before the appearance of clinical findings has become crucial to an early diagnosis and treatment.10,11
FD has been considered to be rare, since its original reported incidence was 1:117,000 for the general population. In males, the reported affection is from 1 in 40,000 to 1 in 238,000 individuals.12,13,14
An accurate estimation of its epidemiology is difficult because FD is clinically very heterogeneous and its symptoms tend to be non‑specific and often unrecognized.15 However, in certain populations, a greater incidence has been described: 1 in 1,250 births for late‑onset disease and 1 in 37,000 births for classic phenotype.16,17,18
The frequency of FD is obviously higher in high‑risk populations, such as those with left ventricular hypertrophy (LVH), stroke and kidney failure requiring dialysis.
Several studies report frequencies of hemodialysis patients affected by FD ranging from 0.11% to 1.2%.3,19,20,21,22
The aims of this study were to search for GLA gene changes in our population submitted to dialysis (hemodialysis or peritoneal); to develop the corresponding genealogic tree with a complete family study; to establish which patients have a pathogenic mutation with a definitive diagnosis of FD, and to assess the clinical indication for ERT.
METHODS
The study was initially approved by the ethics committee of the local hospital, and written consent was obtained from the patients before the samples were collected. Patients who expressed the desire not to participate or those whose inclusion was not deemed possible due to their clinical status at the judgment of the physician in charge of their treatment, were excluded from the study. Enzymatic activity of α‑Gal A and control enzymes acid sphingomyelinase and β‑glucoronidase were determined in dried blood spots (DBS).
Four blood drops from a finger prick were directly applied onto a filter paper (filtercard) with 10 spots completely filled, corresponding approximately to 0.5 ml of blood. It was allowed to dry at room temperature and stored at 2-4 °C before being centrally processed by Centogene AG®. After adding an enzymatic substrate to DBS samples and allowing the proper incubation period to pass, the micromoles of hydrolyzed substrate per liter of blood per hour, units by which GLA activity is expressed, were then calculated through fluorometry.23 α‑Gal A activity below the threshold of 2.6 μmol/L/h was defined as a positive result. Concentration of the biomarker lyso‑Gb3 was determined in the DBS by liquid chromatography mass spectrometry (expressed in ng/mL).24 The normal range was considered 0.00-1.99 ng/mL. The sequencing of the GLA gene was performed by the same laboratory using an amplicon‑based next‑generation sequencing approach. The amplicons cover the entire coding region and the highly conserved exon‑intron splice junctions, with a minimum coverage of >20x for every amplicon.
Missing regions or regions of poor quality were completed with classical Sanger sequencing to achieve 100% coverage. The databases used for GLA variant annotation were the Human Gene Mutation Database (HGMD), Genome Aggregation Database (gnomAD), Exome Sequencing Project (ESP), 1000Genome project (1000G) and CentoMD®.
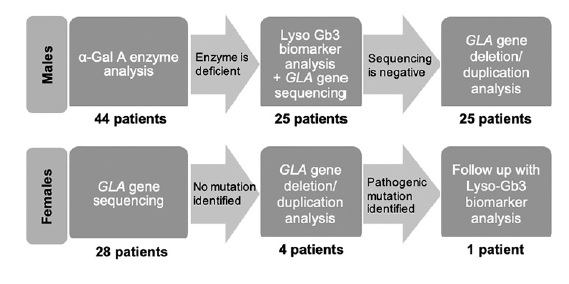
In male patients with low α‑Gal A activity, a genetic study of the GLA gene was performed and in those with normal enzyme activity FD was excluded. Heterozygotic females cannot be reliably defined by enzymatic analysis since their α‑Gal A activity can range from the low level found in affected males to normal level. Therefore, genotype was performed for all of our female subjects (Figure 1).
Estimated glomerular filtration rate (eGFR) was determined by the Chronic Kidney Disease Epidemiology Collaboration (CKD‑EPI) formula in adults and Schwartz equation in children.
Statistical analysis was mainly descriptive. Nominal variables were described using frequency tables, and continuous variables were described using central tendency and dispersion measures. Pooled prevalence of individuals with a GLA gene variant was calculated. The level of significance used was α = 0.05 (two‑sided). All data was analysed using SPSS Statistics® version 23.0.
RESULTS
Two patients refused to participate and 10 were excluded on clinical criteria. A total of 72 patients under dialysis, from a single center, were screened. Of these, 44 (61.1%) were male, and 28 (38.9%) were female, with a median age of 60.5 ± 29 years (range 19-86). Eleven patients, corresponding to 15.3% of the study population, were undergoing peritoneal dialysis (PD). In males submitted to renal replacement therapy (RRT), median α‑Gal A activity level was 2.3 ± 18.2 μmol/L/h (range 0.7‑39.3). Of these, 56.8% (N = 25) had low α‑Gal A activity and were submitted to genetic testing. On the other hand, sequence analysis was carried out in all women, with a total of 53 patients genetically screened.
The overall prevalence of GLA gene variants was 5.6% (N=4). Two of these patients were under hemodialysis (HD), representing 3.2% of this population and 2 were under peritoneal dialysis (PD), 18.2% of PD screened patients (Tables 1 and 2). The prevalence of a pathogenic genetic variant in the GLA gene was 2.8% (N=2) in our cohort.
Table 1 Patients with a GLA genetic change.
| Patient | Mutation | Gender | Age (years) | α-Gal A activity (reference range > 2.6 μmol/L/h) | LysoGb3 (reference range < 1.99 ng/mL) |
| 1 | p.D313Y | Female | 60 | - | 1.3 |
| 2 | p.R118C | Female | 48 | - | 1.6 |
| 3 | p.M290I | Female | 37 | - | 1.4 |
| 4 | p.T194A | Female | 51 | - | 4.0 |
Table 2 Characteristics of positive screened patients
| Case | 1 | 2 | 3 | 4 |
| Age at onset | 54 | 43 | 27 | 50 |
| Type of RRT | PD | HD | PD | HD |
| Etiology of RF | Unknown | Unknown | Unknown | Unknown |
| Medical background | Arterial hypertension | Arterial hypertension | Pre-eclampsia | Arterial hypertension |
| Referral to nephrology department | Referred with stage 5 CKD and started RRT the same week. | Referred with stage 4 CKD and 4 years later initiated RRT. | Referred with stage 3 CKD and subnephrotic proteinuria. Started RRT 7 years later. | Referred with stage 3 AKI and nephrotic range proteinuria. Started RRT the same week without recovering kidney function. |
| Clinical findings | None | Supraventricular tachycardia; LV hypertrophy; MRI T2/FLAIR hypersignal areas in the frontoparietal white matter of vascular etiology. | LV hypertrophy; Hypohidrosis; Heat intolerance. | Corneal opacities; Angiokeratoma; Lacunar cerebral infarcts (CT). |
| Mutation | p.D313Y | p.R118C | p.M290I | p.T194A |
| Diagnosis of Fabry Disease* | No | No | No | Yes |
| Follow-up | No | No | Yes | Not maintained as the patient suffered sudden death |
Case 1 was a carrier of a heterozygous variant c.937G>T (p.Asp313Tyr; D313Y) in exon 6 of GLA gene. Family screening was performed in 2 relatives, with 1 positive result (Figure 2).
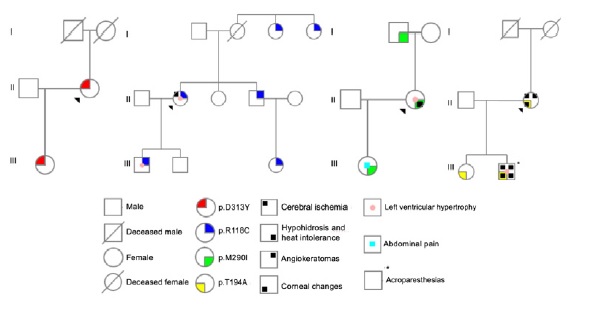
In case 2, n heterozygous variant c.352C>T (p.Arg118Cys; R118C) in exon 2 of GLA gene was found. Among the 7 relatives tested, 5 had the same mutation (Figure 2).
The third case had a heterozygous mutation c.870G>C (p.Met290Ile; M290I) in exon 6 of the referred gene, also found in 2 of the 3 family members tested (Figure 2). All the genetic changes in cases 1 to 3 were previously reported in literature.
In the last case, the proband had a heterozygous mutation c.580A>G (p.Thr194Ala; T194A) in exon 4 of GLA gene, not previously described. The same mutation was present in both the relatives screened (Figure 2).
Family screening was performed in a total of 17 subjects, 9 males (52.9%) and 8 females (47.1%), with a mean age of 35.8 ± 20 years (range 13‑73). Median α‑Gal A activity was 13 ± 10.3 μmol/L/h (range 0.8‑27.7) in males tested. The prevalence of a GLA gene mutation in the family study was 58.8% (Table 3)
Table 3 Family screening results and positive characteristics of the subjects.
| Family | Patient | α-Gal A | Lyso-Gb3 | |||
|---|---|---|---|---|---|---|
| Mutation | (reference range > 2.6 μmol/L/h) | (reference range < 1.99 ng/mL) | Age (years) | Gender | Clinical History | |
| 1 p.D313Y | 1.1 | - | 1.1 | 23 | Female | Drug addiction and attempted suicide in 2010; eGFR 97.3 ml/min/1.73m2, without proteinuria. |
| 2 p.R118C | 2.1 | 7.5 | 1.2 | 22 | Male | Bilateral hydrocele at childhood; Exercise-associated mild LV hypertrophy; eGFR 98.9 ml/min/1.73m2, without proteinuria. |
| 2.2 | 7.2 | 1.4 | 46 | Male | Occupational medicine: normal echocardiogram; eGFR 78.4ml/min/1.73m2, without proteinuria. | |
| 2.3 | - | 1.3 | 23 | Female | Asthma; eGFR 114.2 ml/min/1.73m2, without proteinuria. | |
| 2.4 | - | 1.5 | 67 | Female | Without relevant personal history. | |
| 2.5 | - | 1.4 | 65 | Male | Arterial hypertension; eGFR 77.4 ml/min/1.73m2. | |
| 3 p.M290I | 3.1 | 2.8 | 1.7 | 73 | Male | Osteoporosis; Arterial hypertension; eGFR 76.2 ml/min/1.73m2 without proteinuria; Mild LV hypertrophy. |
| 3.2 | - | 1.3 | 12 | Female | Abdominal pain of unknown etiology; eGFR 96 ml/min/1.73m2 without proteinuria. | |
| 4 p.T194A | 4.1 | 0.8 | 59.2 | 20 | Male | Cornea verticillate; Angiokeratomas; LV concentric hypertrophy; Acroparesthesias; Hypohidrosis; Heat intolerance; Pulvinar Signal (brain MRI); Renal Failure: *5y: arterial hypertension and nocturnal enuresis *8y: CKD probably due to vesicoureteral reflux *12y: started RRT *21y: kidney transplantation |
| 4.2 | - | 6.2 | 28 | Female | Without relevant personal history. |
eGFR - estimated glomerular filtration rate; LV - left ventricle; MRI - magnetic resonance imaging; PD - peritoneal dialysis; HD - hemodialysis; IVS - interventricular septum; y - years old; CKD - chronic kidney disease; RRT - renal replacement therapy.
DISCUSSION
p.D313Y was originally reported in 1993 by Eng et al., in a German male with a classic phenotype.25 However, the presence of a second novel missense mutation, p.G411Y (c.1232G > A in exon 7) was revealed with further analysis.26 Thus, it was crucial to determine whether p.D313Y was a sequence variant or a disease‑causing lesion. Yasuda et al. concluded that p.D313Y mutation is a sequence variant that results in about 60% of wild‑type activity in cells, but lower α‑Gal A activity in plasma. This phenomenon, called pseudodeficiency, is due to mutant enzyme activity being pH dependent: relatively stable at acidic pH, but unstable at neutral pH. On the other hand, the presence of p.D313Y substitution with another mutation on the same allele may further impair the α‑Gal A glycoprotein’s activity or stability.27,28
D313Y enzyme was analysed in patients with cerebrovascular disease, a risk group for FD. In the Belgian Fabry Study (BeFaS) 1.1% of the studied population were found to carry the p.D313Y mutation, consistente with the PORTYSTROKE study findings of 1.2% prevalence of this allele.29,30However, the prevalence of this variant in the normal population is not clear since it was reported in 0.45% of normal X‑chromosomes but could not be found in 415 healthy Portuguese control subjects.27,30 Newer studies doubted the pathological significance of this mutation as there was no evidence of lysosomal substrate accumulation in the tissues expressing them. Several experts reported p.D313Y as a likely benign, non‑pathogenic polymorphism, included in the so‑called Class 2 mutations.10,27,28,31,32,33
The pseudodeficient α‑Gal A activity in plasma and p.D313Y allele low frequency in the normal Caucasian population may lead to the misdiagnosis of FD. Similarly to p.D313Y, the pathogenicity of the exonic variant p.R118C has been controversial. The PORTYSTROKE study identified 3 males and 3 females carrying the R118C enzyme. However, in its post‑hoc epidemiological assay, a case‑control analysis was performed between all cohort patients aged 45 years or less and healthy adult bone marrow donors used as gender and age‑matched controls. The estimated odds ratio for the risk of stroke among carriers of p.R118C did not reach statistical significance.30 This genetic variant was also identified as a pathogenic mutation in screened patients among high‑risk populations for FD. However, the studies failed to provide convincing evidence that the clinical changes found were attributed to FD beyond reasonable doubt.34,35,36
More comprehensive and critical studies37,38showed that the mild deficiency of α‑Gal activity associated with p.R118C is not of enough magnitude to cause major complications of FD. The assumption that it is a pathogenic mutation causing a later‑onset FD phenotype was based on theoretical considerations and p.R118C is most likely a non‑pathogenic or of low‑pathogenicity exonic variant.18,39,40
p.M290I was first reported as a mutation related to the classic form of FD;41 however it is poorly described in the scientific literature. Apart from being found in a Brazilian screening of individuals undergoing HD, in a Portuguese study of hypertrophic cardiomyopathy (HCM) patients and in a Spanish newborn screening, there is no provided evidence of Gb3 and/or LysoGb3 accumulation.42,43,44,45
Thus, the lack of evidence of disease activity suggests that the p.M290I mutation is, at most, mildly pathogenic. p.T194A is a new mutation, not previously described in FD databases like the Human Gene Mutation Database (HGMD), International Fabry Disease Genotype‑Phenotype Database (dbFGP), the Universal Protein Resource (UniProt), Genome Aggregation Database (gnomAD), Exome Sequencing Project (ESP), 1000Genome project (1000G), CentoMD® and the International Genome Sample Resource (IGSR).
In all patients, the mutation was discovered during the screening, with no apparent previous clinical suspicion of FD. The four cases have in common a late referral to nephrology, which made it impossible to perform a biopsy and obtain a definitive diagnosis of the kidney disease etiology. Hypertensive nephroangiosclerosis can be assumed to be the cause of kidney disease in cases 1, 2 and 3. Patients 2 and 3 had other changes that can be found in cases with long‑standing hypertension, such as ventricular hypertrophy and/or ischemic brain injuries.
The patient with the p.T140A genetic variant started dialysis in the context of non‑recovery of an AKI. It can also be assumed that the acute injury was a progressive event of a chronic kidney disease already present, associated with high blood pressure.
In our study, the prevalence of FD among dialysis patients was higher than previously reported. However, there are several limitations; namely the small size of our cohort, the uncertainty about the pathogenicity of some mutations and the putative etiology of the kidney disease in the four patients with a GLA genetic variant. The recognition of the novel mutation p.T194A is important for better knowledge of FD and its spectrum of clinical manifestations. Patients carrying this mutation are expected to develop a classic and life‑threatening FD phenotype. Two new cases were also determined as a result of family screening, for which follow‑up and FD treatment can be a future option.
Screening of a dialysis population with a dried blood spot test is a simple procedure and might identify patients who remain otherwise unrecognized. Even so, since there are no guidelines for successful and beneficial screening, it should be carried out with great caution.
A clinical correlation to assess organ involvement and to identify FD manifestations is crucial to avoid a false diagnosis. It is also importante to establish a correct pedigree and screen relatives in whom progression of the disease can be detected at an earlier stage.
We encourage screening programs among patients with end‑stage renal disease, in order to possibly identify new cases of FD, provide specific clinical care and eventually initiate ERT.

