











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Systemic lupus erythematosus (SLE) and ANCA-associated vasculites (AAV) are traditionally considered to be two distinct conditions that affect the kidney structurally and functionally through diferente mechanisms1-3.
SLE is characterized by the presence of serum autoantibodies against nuclear components such as antinuclear antibodies (ANA), extractable nuclear antigens (ENA) antibodies, which include RNP and Smith antigens, and double strand DNA (dsDNA) antibodies1-3.
It has a heterogeneous clinical presentation, displaying a wide spectrum of manifestations including lupus nephritis (LN) in 30-60% of cases1-4and vasculitis in 11% of patients5. SLE leads to an imune complex glomerulonephritis. The patterns of glomerulonephritis seen in LN have been classified by the 2004 International Society of Nephrology (ISN) /Renal Pathology Society (RPS) classification (GN)6,7.
On the other hand, AAV is a small vessel systemic vasculitis caused by serum autoantibodies against polymorphonuclear leucocytes, most commonly anti-neutrophil cytoplasmic antibodies (ANCA) anti-myeloperoxidase (MPO) or anti-proteinase 3 (PR3)3 and is expressed in the kidney as pauci-immune crescentic glomerulonephritis1,8,9. Vasculitis may occur in patients with SLE, but rarely meets the criteria for AAV3.
The SLE/AAV overlap syndrome (SLE/AAV OS) was first described in 20084-7 for patients who met criteria for both conditions. It seems that a possible shared immune pathogenesis may exist, but the exact mechanism is unclear2 and that the overlap of SLE and AAV may be part of a poliautoimmune syndrome suggested by association with a third autoimmune disease in some patients.
CASE REPORT
A 47-year-old African-descent woman with an unremarkable past medical history presented with nephritic syndrome and severe rapidly progressive renal failure requiring renal replacement treatment (RRT).
She complained of asthenia and significant weight loss over the previous 6 months.
The laboratory tests showed a serum creatinine (sCr) of 8.2 mg/dL, microscopic hematuria and proteinuria of 3.9g/day. The immunological workup identified MPO-ANCA (6.63 UI/mL), ANA (titer >1/640) and nRNP/Smith antibodies. Other specific antibodies were absent. The kidney biopsy showed a crescentic necrotizing pauci-immune GN, with cellular and fibrocellular crescents in 55% of the glomeruli (Figure 1A) with tubuloreticular inclusions in glomerular endothelial cells (Figure 1B) and rare mesangial electrodense deposits on electron microscopy (EM).
She was diagnosed with MPO-AAV and started induction treatment with pulsed methylprednisolone (1g) for 3 days followed by prednisolone and IV cyclophosphamide (CyP) 750 mg/m2 monthly. Additionally, she was on plasma exchange treatment for 6 sessions. There was a favorable response after the first CyP cycle, with improvement of kidney function and RRT suspension. Complete remission was achieved after the third month of treatment. She received an additional 2 months of IV CyP but due to an increase of liver enzymes, CyP was withdrawn, and maintenance treatment with azathioprine (AZA) (50mg q.d.) and prednisolone was started. However, after four months, AZA was also withdrawn for the same reason. Other causes for the elevation of the liver enzymes, such as hepatotropic viruses, were excluded and liver ultrasound showed no alterations. Prednisolone was tapered over 6 months, completing a total of 15 months. She maintained complete AAV remission through long-term outpatient nephrology follow-up, but ANA antibodies remained positive.
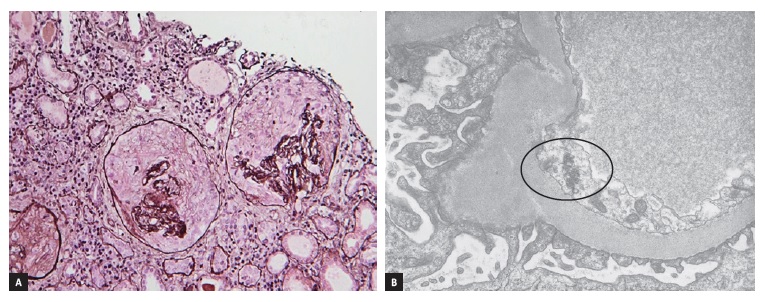
Figure 1: (A) Two glomeruli with cellular crescents; residual tuft collapse with no endocapillary proliferation (Methenamine Jones Stain, x10). (B) Electron micrograph showing tubuloreticular inclusions in glomerular endothelial cells (black circle, x10000)
Five 5 years later she presented with non-nephrotic proteinuria (2g/day), microscopic hematuria and tenderness and morning stiffness on several joints. A new immunological workup showed persistency of ANA (>1/640) and nRNP/Smith antibodies and de novo dsDNA (54UI/ml), sp100 and gp210 antibodies. Complement factor C3 levels were decreased and C4 levels were within normal range. ANCA antibodies were negative. Given these findings, she was diagnosed with SLE and initially treated with hydroxychloroquine (HCQ) and angiotensin converter enzyme inhibitor (ACEI) for 10 months. However there was na increase of proteinuria with persistency of all serum antibodies and a new kidney biopsy was performed.
The second kidney biopsy showed a diffuse mesangioproliferative GN (Figure 2A), with sclerotic lesions in 64% of glomeruli on light microscopy. There was no significant interstitial inflammation, interstitial fibrosis or tubular atrophy. Immunofluorescence (Figure 2B) showed small IgG granular deposits in the mesangium and in the capillary wall. Staining for IgA, IgM, C3, C4 and C1q was negative. EM showed focal endocapillary proliferation and amorphous electrodense deposits in the sub-endothelial compartment (Figure 2C) and in the mesangium. There was also persistency of tubuloreticular inclusions. Given the clinical context, these findings were consistent with lupus nephritis class III with a NIH activity index of 1/24 and chronicity índex of 3/12). She started treatment with pulsed methylprednisolone (250mg) for 3 days, followed by prednisolone and mycophenolate mofetil (MMF) (500mg b.i.d.). However, a progressive increase of liver enzymes determined the withdrawal of MMF after 3 months. She maintained treatment with HQC, ACEI and prednisolone, which was tapered and discontinued after 12 months, with progressive improvement of kidney function and proteinuria and partial serologic remission, with ANA persistency.
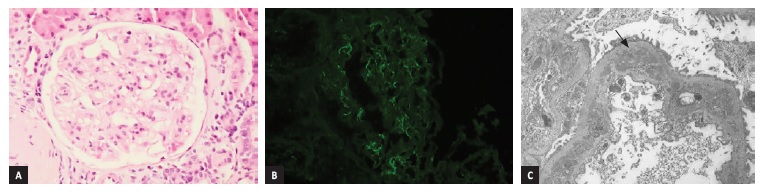
Figure 2: (A) Light mesangial matrix expansion with segmental hypercellularity (H&E, 20x). (B) Immunofluorescence with small and rare IgG deposits in the mesangium and in the capillary wall (IgG, 20x). (C) Electron micrograph showing electrodense deposits, in sub-endothelial compartment, with remodeled basement membrane and podocyte foot process effacement (black arrow) (x5000)
Given the recurrent elevation of transaminases, alkaline phosphatase and gamma-glutamyl transferase and the presence of anti-Sp100 and anti-gp210 antibodies along with ANA, she was later diagnosed with primary biliary cholangitis (PBC), and was later started on ursodeoxycholic acid. Liver elastrography showed mild to moderate
liver fibrosis (F1-F2).
DISCUSSION
This case report describes a challenging diagnosis of SLE/AAV OS as part of poliautoimmunity syndrome, as it was associated with the diagnosis of a third autoimmune disease during the long-term follow-up of the patient. SLE/AAV overlap syndrome (SLE/AAV OS) was first described in 2008 by Nasr and colleagues4-7for patients who met criteria for both conditions. The prevalence of SLE/AAV OS is low, representing about 2% in patients with GN as published in a 2016 French national survey10.
Most of the patients are female, with a mean age around the second decade, showing severe clinical features at presentation such as pauciimmune necrotizing GN and pulmonary hemorrhage11. AAV and SLE can develop concurrently or several years apart and features like the presence of ANA and tubuloreticular inclusions at the time of AAV diagnosis such as in the present case are common11. The vast majority of cases reported are MPO-ANCA7.
There is some evidence in the literature of an association between SLE and AAV2. SLE and AAV share some immunological features and can probably be triggered by the same stimuli. Also, some authors suggest that crescentic necrotizing pauci-immune GN may represent a rare variation of LN, in which ANCA is the driving factor via neutrophil
activation, cytokine release and endothelial damage, resulting in necrosis and crescent formation2,12,13. On the other hand, ANCA antibodies frequently occur in SLE patients (16-20%) but many these patients do not develop vasculitis9. ANCA seropositivity is even higher in patients with SLE with renal involvement and the occurrence of both diseases simultaneously seems to occur more frequently than just by coincidence2,10-12.
Our patient presented initially with MPO-AAV, classified as microscopic polyangiitis according to the 2012 Chapel Hill classification14.
She was successfully treated with immunosuppression using the CYCLOPS pulsed IV cyclophosphamide regime15 and plasmapheresis, with complete renal function recovery and serological remission. Even though there were ANA and RNP antibodies at the time of AAV diagnosis, she had no other SLE defining criteria.
Regarding the SLE diagnosis 5 years later, she scored 25 points on the new 2019 EULAR/ACR criteria for a minimum of 10 points for SLE diagnosis. These included the presence of ANA antibodies and joint involvement with tenderness and morning stiffness on more than two joints (6 points); presence of dsDNA and Smith antibodies (6 points); decreased C3 serum levels (3 points), and biopsy-proven class III Lupus Nephritis (10 points)16. She also scored 5 criteria on the 2012 Systemic Lupus Collaborating Clinics (SLICC) criteria17 for a minimum of 4 for SLE diagnosis, one of them being a clinical criterion with biopsy proven class III Lupus Nephritis (2004 International Society of Nephrology (ISN) /Renal Pathology Society (RPS) classification)6 and the remaining, immunological criteria (presence of ANA, dsDNA and anti-Smith antibodies and low C3 levels).
The clinical presentation, serological pattern and kidney biopsy findings determined the start of immunosuppressive treatment, with a positive response. The integration of light microscopy, IF and EM, allowed a more accurate diagnosis on the second biopsy18. Although IF had been scarce, the presence of mesangial and sub-endothelial deposits, the tubuloreticular inclusions and overall clinical contexto supported LN and SLE diagnosis.
Adding to the complexity of this case was that the patient was diagnosed with PCB. The association of this condition with SLE or AAV is rare and, to the best of our knowledge, the association with SLE/AAV OS has not been previously described in the literature. One review regarding SLE and PBC in 34 case reports concluded that most of these patients were female, and PBC was usually diagnosed before SLE, with SLE being late onset19. Another review, published in 2019, showed that the association of AAV with other autoimmune disorders is rare; only 3 out of 28 patients with AAV associated to another autoimmune condition presented with AAV and PCB simultaneously20. According to some authors, SLE and AAV could be part of a complex poliautoimmune syndrome, in which SLE/AAV OS can be associated with a third autoimmune disease, such as the present case10. Tissue damage during an immune response can lead to the priming of self-reactive T and/or B lymphocytes, despite the initial insult. The shift and diversification of autoantibody targets during the course of autoimmune diseases has been defined as epitope spreading21 and has been described in SLE, explaining the broad of autoantibodies that can be found in this disease and also the association with other autoimmune diseases.
This phenomenon was also studied in patients with AAV and other autoantibodies or other autoimmune diseases22.
Given the diagnosis of focal proliferative LN, this patient has a high risk for relapse and progression to chronic kidney disease and according to current guidelines she should receive maintenance treatment with a second immunosuppressive drug in addition to prednisolone, such as MMF or AZA. However, regarding the PCB diagnosis, the patient has mild to moderate liver fibrosis and serologic markers, such as ANA, anti-Sp100 and anti-gp210 that correlate with progression to chronic liver failure and cirrhosis. Also, it is likely that the immunosuppressive drugs contributed to the transitory elevation of liver enzymes on the various occasions. In a trial from 2005, the use of MMF on PCB patients was associated with a high percentage of adverse advents23. In the future, other therapeutic approaches for LN, such as rituximab24, should be considered.

