











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Antineutrophil cytoplasmic autoantibodies (ANCAs) are associated with ANCA-associated vasculites.1,2Neutrophil activation by ANCA induces endothelial injury with vascular inflammation. (1) The typical histological-pattern in the kidney is pauci-immune necrotizing and crescentic glomerulonephritis (GN).3,4
Even though recent studies highlight the importance of kidney biopsy for the diagnosis and prognosis of ANCA-associated GN, (4)(5) some classifications, such as the European Medicine Agency Classification,6 dismiss kidney biopsy. In this classification, ANCA-associated GN is defined by hematuria7 associated with red cell casts or with proteinuria with positive ANCA serology.6
A positive antiproteinase 3 (PR3) or antimyeloperoxidase (MPO) in a patient with suspected glomerulonephritis has >95% association with ANCA-associated GN, (7) but these autoantibodies are not pathognomonic for this disease and can appear in other conditions.
Healthy individuals can have circulating autoantibodies against MPO and PR3.8ANCA, specially MPO, can also occur in a wide range of other settings, e.g. lupus nephritis, endocarditis, other chronic infection, malignancy or associated with drugs.5
The pathogenesis of drug-induced ANCA-associated vasculitis might be multifactorial and the diagnosis is based on the temporal relationship between clinically evident vasculitis and administration of the offending
drug, after exclusion of other medical conditions.9 This diagnosis requires an accurate investigation and high clinical suspicion.
We present a case of a patient with high titers of MPO-ANCA and rapidly progressive kidney dysfunction, hematuria, and proteinuria. Kidney biopsy showed surprising findings with different treatment and prognosis from ANCA-associated GN. This case report praises the importance of kidney biopsy, and points out to other causes of high ANCA titers.
CASE REPORT
A 60-year-old white male presented to the emergency department (E.D.) with generalized pitting edema and decreased urine output.
One month before admission, the patient started gaining weight (about 10 kg) accompanied by peripheral edema and a non-pruritic erythematous rash. One week earlier he had noticed decreased urine output. He reported no fever, constitutional, uremic, or respiratory symptoms. He also denied other complaints.
His past medical history included benign prostatic hyperplasia, lumbar herniated disc, dyslipidemia and he had recently been diagnosed with depression. His usual medication was tamsulosin (0.4 mg id), pregabalin (150 mg id), rosuvastatin (5 mg id) and clozapine (25 mg id). The latter was introduced two months before hospital admission.
There was no history of other recent medications, drugs, or herbal medicine. Previous laboratory results showed serum creatinine (sCr) of 1.28 mg/dL.
On physical examination at E.D., he was hypertensive (blood pressure 155/88 mmHg) and afebrile. He presented generalized pitting edema and a diffuse erythematous rash on both legs, arms and chest with no hand or foot involvement. The remainder of the physical examination was unremarkable.
The initial laboratory test results revealed elevated serum creatinine of 2.1 mg/dL and urea of 65 mg/dL. He had also dysproteinemia with hypoalbuminemia (2.3g/dL) and dyslipidemia (total cholesterol 240 mg/dL and triglycerides 177 mg/dL). Urinalysis revealed specific gravity of 1.014, albumin 4+, erythrocytes 5-10 cells per high-powered field (HPF) and leukocytes 2-3/HPF. Urine protein/creatinine ratio of 11g/g. No cytopenias or elevated inflammatory markers were found. Uric acid was 6.4 mg/dL. Thyroid function and B-type Natriuretic Peptide (NT-proBNP) were normal. Other laboratory parameters were unremarkable.
Renal ultrasonography revealed normal appearing kidneys, without obstructive uropathy. Lung X-ray was unremarkable. The patient was admitted to the Nephrology Department where he started diuretics and antihypertensive drugs. He maintained statins. Clozapine was discontinued since it was the last drug to be introduced.
A high titer (111 UI/ml (RR<5) of myeloperoxidase antibodies (MPOANCA) was found. Remaining autoimmune studies were negative, including serum complement fractions and immunoglobulins. Serum protein electrophoresis test and immunofixation were normal. No eosinophilia or eosinophiluria was detected. Rheumatoid factor, cryoglobulins and viral serologies were negative.
Since admission, his serum creatinine rose, reaching a maximum of 4.9 mg/dL after 10 days of admission (Graphic 1). Pulses of methylprednisolone (500 mg 3 days) were started.
In summary, we had a patient with acute kidney injury, skin rash, hematuria, and a nephrotic syndrome. The only positive laboratory workup finding was a high titer of MPO.
Kidney biopsy was performed after six days of admission, and we obtained 19 glomeruli, one of which was globally sclerosed. The remaining showed slight increase in diameter and segmental expansion of the matrix accompanied by mild endocapillary and mesangial hypercellularity.
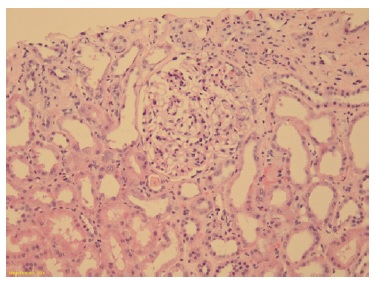
Podocytes were hyperplasic and occasionally containing protein granules. Proximal and distal tubules presented degenerative and reactive changes, with cell debris in the lumen (Figure 1 and 2). Rare apoptotic bodies were observed in the epithelial tubular stroma with edema and mild inflammatory infiltrate. Arterioles were well preserved. Congo red stain was negative. Immunofluorescence was negative for immunoglobulins, complement fragments, albumin, and fibrin. Electron microscopy was performed using formalin-fixed material and demonstrated markedly swollen podocytes with foot process effacement. No electron dense deposits were found (Figure 3).
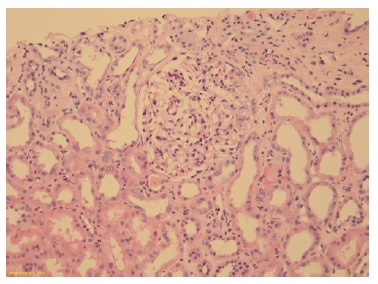
Figure 1: Glomeruli showing segmental mesangial hypercellularity and endocapillary proliferation with leucocytes. Tubules with epithelial injury with thinning and loss of apical cytoplasm. (Hematoxylin and eosin 10x)
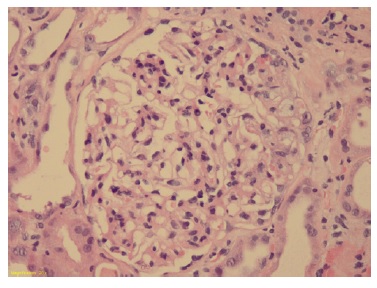
Figure 2: Glomeruli showing segmental mesangial hypercellularity and endocapillary proliferation and hyperplasia of podocytes (Hematoxylin and eosin 20x)

Figure 3: Electron microscopy (2500x, swollen podocyte (arrow) with foot process effacement and no electron dense deposits
In conclusion, focal segmental glomerulosclerosis (FSGS) celular variant with mild acute tubular injury was identified. On the 7th day of admission, the rash disappeared with no specific therapy. After a three-day course of pulsed methylprednisolone, he was treated with 4 weeks of oral prednisolone 1mg/Kg/day followed by slow taper. He achieved clinical and laboratory response after two weeks (sCr 1.6 mg/dL and a proteinuria of 400 mg/24h). The repeat MPO-ANCA titers were negative at one-month follow-up. Presently, the patient is in complete remission one year after suspension of steroid therapy.
„
DISCUSSION
We present a challenging case of a patient with kidney dysfunction, nephrotic syndrome with massive proteinuria, a skin rash and high MPO titer.
Our first diagnosis hypothesis was a pauci-immune GN. Although ANCA GN presenting with nephrotic syndrome is unusual, there are some reported cases, particularly when associated with anti-phospholipase A2 receptor antibodies.10
Unexpectedly, kidney biopsy revealed a diagnosis with a significantly different therapeutic approach and prognosis: cellular variant FSGS with mild acute tubular injury.
Indeed, the levels of MPO and skin rash can be explained by the recent introduction of clozapine. Clozapine is an antipsychotic medication that is used in some cases of major depression.11 Several reviews of drug induced ANCA-associated vasculitis present clozapine as one possible offending agent.9,12
The pathogenesis of drug-induced vasculitis remains unclear and it is thought to be multifactorial. The clinical manifestations are like those of primary ANCA-associated vasculitis. Drug-induced vasculitis usually attacks the skin and sometimes the subcutaneous part of the skin, as in our case, but sometimes also the kidney. The offending drugs and their metabolites may accumulate within neutrophils, which bind to MPO and modify its configuration, spreading the autoimmune response.9
The relationship between the skin rash and high MPO titer with the administration of clozapine, as well as clinical remission and negative MPO after its discontinuation favors this hypothesizes. Unfortunately, a skin biopsy was not obtained to prove this hypothesis. On the other hand, positive MPO-ANCA results can appear in diferente situations, such as infectious, malignant, or autoimmune diseases and even in healthy individuals. In these circumstances, the titers are usually low and do not typically disappear.12
Regardless of the presence of MPO, kidney biopsy did not show vasculitic findings. We documented focal segmental glomerulosclerosis (FSGS) cellular variant with kidney dysfunction. D’Agati et al.13 proposed a pathologic classification of FSGS in 2004 based on light microscopic patterns: the Columbia Classification. This categorization can be applied to all etiologies and presumes exclusion of other glomerulopathies that course with scaring.
According to this, FSGS is subdivided into five variants: not otherwise specified (NOS), perihilar (PH), cellular (CELL), glomerular tip lesions (GTL) and collapsing (COLL) variant.13 Cellular variant is the least common variant, identified in only 3% of cases. This is defined by at least one glomerulus with endocapillary hypercellularity (foam cells, macrophages and other leucocytes, endothelial cells) involving more than 25% of the glomerular tuft. This variant may lack segmental scars, mimicking focal proliferative GN. Foot process effacement is usually severe.14,15Since COLL and CELL variants show epithelial cell proliferation with dysregulated podocyte phenotype, some investigators do not distinguish these two types. Stokes et al. (14) made the first attempt to compare CELL with other FSGS variants (COLL, GTL and NOS). In this study there were significantly different rates of remission and end stage renal disease between the cellular and collapsing variants. This supports that these two variants should not be classified together.
Additionally, in this same study, CELL and GTL showed very similar pathologic findings, suggesting that CELL can represent a more advanced stage of GTL.
The responsiveness of CELL to treatment is intermediate between COLL and GTL. However, other clinical aspects differences are still unclear.15,16Since it is the rarest of the variants, it is also the least understood. The majority of CELL cases present characteristics of nephrotic syndrome with 4-6 months of evolution and a low sCr level (between 1.58 ± 0.18 mg and 2.5 ± 1.7 mg/dl).14,16No reports correlating this variant with drugs, particularly clozapine, were found.
In this case, the patient had shorter evolution of nephrotic syndrome (1 month) and the response to corticosteroid therapy was excellent. Kidney dysfunction in this case was probably due to acute tubular injury, also documented in the histologic findings. Massive proteinuria in a short period of time, associated with hemodynamics changes due to diuretics and antihypertensive drugs, can explain the acute tubular damage seen in the kidney biopsy.
This case report highlights the importance of a comprehensive clinical history, along with kidney biopsy for an accurate diagnosis.
CONCLUSION
Kidney biopsy is still a very important tool for diagnosis and prognosis. Although ANCA autoantibodies can be an essential clue to the diagnosis of ANCA-related GN, kidney biopsy should not be dismissed. This case report emphasizes the importance of biopsy and how diferente the management of a patient with high titer of MPO and kidney dysfunction would be if it were not performed.

