











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Membranoproliferative glomerulonephritis (MPGN) is a rare, chronic glomerulonephritis in pediatric age. The prevalence of MPGN has not been well-established, but it has been suggested to vary depending on age and geographical location.1,2
In children, MPGN is frequently idiopathic, whereas in adults it is commonly associated with secondary causes. A respiratory tract infection may precede the diagnosis in half of the patients.3
Nephrotic syndrome (NS), nephritic syndrome, asymptomatic proteinuria with hematuria, hypertension and often impaired renal function are the clinical features that might be observed at initial presentation. Its incidence in children with nephrotic syndrome is approximately 4-8%.2,4
In the past, MPGN was diagnosed and classified by renal histological features and grouped into three pathological subtypes with diferente etiologies and pathogenesis. Activation of the alternative complemente pathway has repeatedly been observed in conjunction with low sérum levels of complement C3. A link between dysregulation of the alternative complement pathway and the pathogenesis of MPGN was assumed and classification based on pathogenesis has recently been reconsidered, with division into those cases in which the glomerular imune deposits stain for immunoglobulins and complement and those cases that are characterized by C3 deposition alone.4
The prognostic factors, the outcome and the most suitable treatment regimen are incompletely known in children. Children with MPGN usually have a poor prognosis, as renal failure occurs within 10 years in 50% of those affected. Impaired renal function after one year of onset is considered a risk factor for poor renal outcome and end-stage renal disease (ESRD).1,4,5
CASE REPORT/CASE PRESENTATION
A previously healthy 3-year-old boy was admitted with a two day history of prostration, peripheral edema, hematuria and oliguria. The child was fully immunized, and his medical history, as well his family history, was unremarkable. Psychomotor development was normal.
He was observed 10 days before with 5 days of fever, and productive cough, with an epidemiological context of scarlet fever at school 3 weeks earlier. A rapid antigen detection test for group A streptococcus was performed and was negative.
Physical examination revealed bilateral eyelid, ankle, and distal tibia edema and blood pressure of 120/92 mmHg (p>99). Laboratory investigation revealed acute kidney injury (creatinine 123 umol/L and blood urea nitrogen (BUN) 18 mmol/L, hyponatremia and hyperkalaemia (Na+ 132 mmol/L, K+ 5,8 mmol/L), hypoalbuminemia (1,7 g/dL), normochromic normocytic anemia (Hb 11,7 g/L, MCV 79,6 fL, MCC 27,5 pg), c-reactive protein (CRP) 1,78 mg/dL and a low level of C3 (0,83 mg/dL, minimum 0,64 mg/dL) and C4 (minimum of 0,09 mg/dL), that normalized after ten weeks and remained normal in subsequente assessments. Urinalysis revealed nephrotic-range proteinuria (ratio prot/creatU 978 mg/mmol) and microscopic hematuria (>20 erythrocytes per field). Renal ultrasound showed bilateral cortical hyperreflectivity. Anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies and anti-glomerular basement membrane antibody were negative. Serum complement CH50 and immunoglobulin (Ig) levels (IgA, IgG, IgM, IgE) were normal. Antistreptolysin O titer (ASTO) and anti-DNAse B was negative initially and remained negative six weeks later. Cultures of the blood, urine and stool were negative. Cytomegalovirus (CMV) and Epstein-Barr (EBV) serology was compatible with past infection and parvovirus was negative.
On day (D) 5 after admission, he showed a clinical worsening, with significant peripheral edema and nephrotic proteinuria, therefore prednisolone (60 mg/m2/day) was initiated. On D10 of corticosteroids, a renal biopsy was performed, given the severe worsening of renal function during this period, which revealed MPGN with presence of diffuse mesangial hypercellularity, endocapillary proliferation, and focal thickening of the walls of the glomerular capillaries and deposits of IgG, IgM and C3 (Figures1, 2, 3). After this, an extended infectious study was performed with HSV 1 and 2, HIV 1 and 2, hepatitis A, B and C serologies that were negative. EBV and CMV serologies were repeated with the same results. However, CMV DNA was detected by PCR (Polymerase Chain Reaction) in urine and blood (1319 copies). CMV genome at biopsy and CMV inclusions in endothelial cells were not identified. Nevertheless, CMV glomerulonephritis was assumed and intravenous ganciclovir 5 mg/kg every 12h was performed for 5 days followed by a maintenance therapy of oral valganciclovir 30 mg/kg/day, once a day with a reduction in viral load (<250 copies) on D5 of antiviral therapy.
On D21, antiplatelet therapy was started with acetylsalicylic acid and dipyridamole, as well as oral pravastatin. The patient needed several albumin administrations (minimum albumin of 17 g/dL), the last one on D36. Hypertension control required triple anti-hypertensive therapy (amlodipine, propranolol and enalapril). Complete renal recovery occurred on D19 (creatinine 35 umol/L, BUN 4 mmol/L), but nephrotic-range proteinuria was maintained, without hematuria. The patient was discharged on D49 under valganciclovir, oral prednisolone 45 mg/m2 daily, anti-hypertensive triple therapy and antiplatelet drugs.
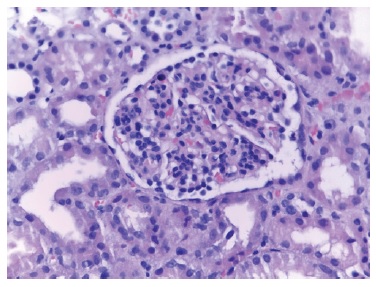
Figure 1 Glomerulus with mesangial enlargement and hypercellularity accompanied by mild endocapillary hypercellularity. Some capillary loops are slightly thickened. HE stain (Hematoxylin-eosin), 400x
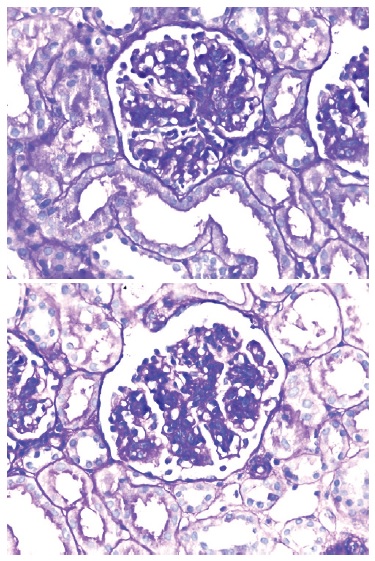
Figure 2 Enlarged glomerulus, with a lobulated appearance, with mesangial enlargement and hypercellularity, and thickening of the wall of some glomerular capillaries with double contours. PAS (Periodic Acid Schiff) staining, 400x.
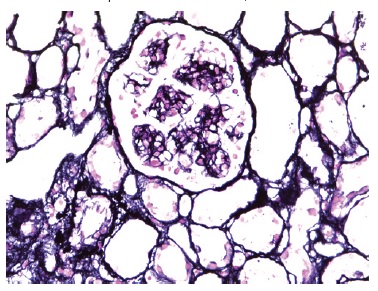
Figure 3 Glomerulus with thickening of the glomerular capillary wall, with some double contours and without spike formation. Marinozzi stain, 400x
At the one-year follow-up consultation, CMV DNA viral load was no longer detectable in serum, but was still detectable in urine. Primary immunodeficiency was excluded. Proteinuria totally resolved after six months without recurrence. Valganciclovir was suspended after six months, and gradual prednisolone weaning was performed over a year. Serial evaluations of renal function were always normal. At the latest follow-up at 14 months, the urine specimen was normal without red blood cells or protein.
DISCUSSION/CONCLUSION
CMV is a ubiquitous virus that commonly infects people of all ages throughout the world. In 90% of the infected individuals, the disease is asymptomatic, while in the remaining 10% CMV infection presents as a mononucleosis-like syndrome. In children, the spectrum of disease caused by CMV infection ranges from asymptomatic or mild disease in immunologically normal hosts to severe and potentially life-threatening disease in newborns and immunocompromised children.6,7
Apart from morbid conditions that may influence cell-mediated immunity, there are no well-known mechanisms predisposing previously healthy people to a severe CMV infection.8
It has been reported that a congenitally infected child, asymptomatic at birth, may develop psychomotor abnormalities and/or deafness later.8,9 In our case, there were no signs or symptoms of these abnormalities, so we assume that he had not had a congenital infection, but rather an acute primary CMV infection or, more likely, a reactivation in a period of immunosuppression secondary to corticosteroid therapy. We did not find cases of membranoproliferative glomerulonephritis caused by CMV in immunocompetent children in the literature. We found a case of a 19-month-old boy with membranous nephritis associated with acquired CMV infection.8
In our case, we were unable to say with certainty that CMV was the etiologic agent, but we still attempted a directed treatment with valganciclovir given the exuberance of the symptoms. Since serologies were never compatible with recent infection and seroconversion did not occur after the initial episode of fever, reactivation of CMV instead of an acute primary infection should be considered. IgM was always negative in our case, so measurement of CMV IgG avidity, to distinguish recent from past infection, could have been useful in our case.10,11,12
In immunosuppressed patients suffering from CMV infection, either symptomatic or asymptomatic, treatment with ganciclovir is recommended. In contrast, because of the treatment’s high toxicity, there are no recommendations for treating immunocompetent individuals, adults and children. Nephrotoxicity and cytopenia due to myelosuppression are among the major side effects of this agent.7 During follow-up, our patient had no adverse effects from the treatment and, therefore, he completed 6 months of valganciclovir. Furthermore, MPGN usually do not show a rapid therapeutic response, as in our case, and need high and prolonged doses of corticosteroid, sometimes in combination with other immunosuppressants.13,14,15
As with other primary glomerulonephritis, pathologic diagnosis of MPGN is based on characteristic histologic findings. MPGN has traditionally been classified according to the locations of electron dense deposits, where MPGN I is characterized by mesangial and subendothelial deposits as we found in our case, or, according to the new classification, as an immune complex-mediated MPGN.
The main causes thereof are autoimmune diseases, infections or idiopathic.14,15,3 Studies in adults and children have shown that proteinuria is a major risk factor for developing ESRD. There is currently no established treatment for MPGN. Patients appear to respond differently to various therapy modalities. The majority of treatment regimens and case series have been reported in adults, not in children, and are often associated with significant side effects.3
In our case, we were not able to attribute a cause for the MPGN, therefore an idiopathic cause is more likely. Although the context of scarlet fever was a plausible etiology, we did not have data in favour: rapid antigen test for group A streptococcus was negative, ASTO and antiDNAse B were never elevated and C3 and C4 remained low longer than expected in post-streptococcal glomerulonephritis. Although there are reported cases of MGPN following a streptococcal infection, this is not likely given the course of our investigation. CMV was detected by PCR, leading us to treat it as if it were the underlying cause, but we cannot establish this with certainty since we did not find genetic material in the biopsy. Regarding treatment, it was kept longer than expected considering an idiopathic etiology, since it usually has a faster therapeutic response to corticosteroid therapy.
In conclusion, MPGN is a rare presentation of nephropathy in children, and treatment, despite not being established, should never be delayed given the risk of ESRD.

